Date: Tue, 11 Apr 2017 11:42:42 +0200
Dear Amber users, Dear Ross Walker,
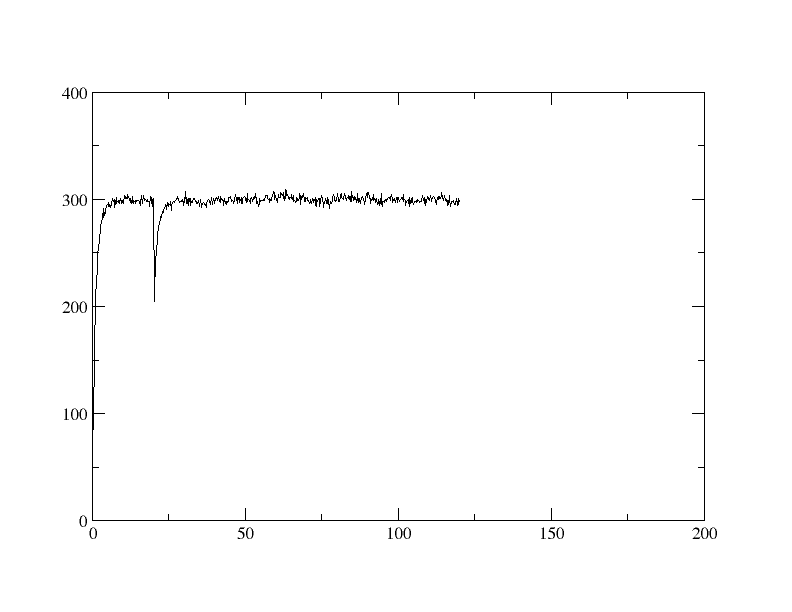
I am running Amber-Tutorial-B1 (DNA) of Ross Walker.
I'm am taking his input-files from Section 5:
http://ambermd.org/tutorials/basic/tutorial1/section5.htm
and run the commands as written on the website, but the Temperature
dropped from 300K down to about 200K (See attachment) when (immediately
after) changing from one Input-File to the next, which shouldn't. (tempi
= 300.0, temp0 = 300.0,)
I uploaded my files to:
http://jokalliauer.bplaced.net/live%20access/Internet/UNI/PhD/B1Win/
(Alternative everything as ZIP):
http://jokalliauer.bplaced.net/live%20access/Internet/UNI/PhD/B1Win.zip
I run the problem with AmberTools16.21 on Cygwin and on Ubuntu 16.04,
both had the same problem, as mentioned above.
I don't know what I did wrong, because I just downloaded the inputfiles
(polyAT_wat_min1.in
<http://ambermd.org/tutorials/basic/tutorial1/files/amber_input_files/polyAT_wat_min1.in>,
polyAT_wat.prmtop
<http://ambermd.org/tutorials/basic/tutorial1/files/amber_input_files/polyAT_wat.prmtop>,
polyAT_wat.inpcrd
<http://ambermd.org/tutorials/basic/tutorial1/files/amber_input_files/polyAT_wat.inpcrd>,
polyAT_wat_min2.in
<http://ambermd.org/tutorials/basic/tutorial1/files/amber_input_files/polyAT_wat_min2.in>,
polyAT_wat_md1.in
<http://ambermd.org/tutorials/basic/tutorial1/files/amber_input_files/polyAT_wat_md1.in>,
polyAT_wat_md2.in
<http://ambermd.org/tutorials/basic/tutorial1/files/amber_input_files/polyAT_wat_md2.in>)
did not edit anything at all and run the commands as written in the
tutorial (Sec5.sh
<http://jokalliauer.bplaced.net/live%20access/Internet/UNI/PhD/B1Win/Sec5.sh>).
Maybe because I use a newer version of AmberTools (16.21)?
According to Amber16-Manual the restart-file should contain all
velocities (therefore also the kinetic energy/temperature?), therefore I
don't understand why it is not working as written in the tutorial.
Best regards from Vienna
Johannes Kalliauer
TU Wien (Vienna University of Technology)
-------- Weitergeleitete Nachricht --------
Betreff: Re: Poly(A)-Poly(T) DNA Tutorial Query
Datum: Thu, 23 Mar 2017 18:32:21 -0400
Von: Ross Walker <ross.rosswalker.co.uk>
An: Johannes Kalliauer <johannes.kalliauer.tuwien.ac.at>
Hi Johannes,
Yes AMBER 16 updated the default restart format to netcdf binary. The
tutorial needs updating. I'll make that modification now. Thanks for
pointing that out.
With regards to the prmtop the file format definition is here:
http://ambermd.org/formats.html
It also has info for the coordinate file etc.
Hope that helps.
All the best
Ross
> On Mar 23, 2017, at 12:37, Johannes Kalliauer
> <johannes.kalliauer.tuwien.ac.at
> <mailto:johannes.kalliauer.tuwien.ac.at>> wrote:
>
> Dear Ross Walker,
>
> First thank you very much for the Amber-Tutorials. They are really
> awesome.
>
> I have a comment to Section4 of the B1-DNA-Tutorial:
> http://ambermd.org/tutorials/basic/tutorial1/section4.htm
> Using the command
>> $AMBERHOME/bin/sander -O -i polyAT_gb_init_min.in -o
>> polyAT_gb_init_min.out -c polyAT_vac.inpcrd -p polyAT_vac.prmtop -r
>> polyAT_gb_init_min.rst
> leads in my case (Amber16) to a binary restartfile.
>
> According to
> http://archive.ambermd.org/201603/0168.html
> I had to modify the code for ambpdb from
>> $AMBERHOME/bin/ambpdb -p polyAT_vac.prmtop *<* polyAT_gb_init_min.rst
>> > polyAT_gb_init_min.pdb
> to
>> $AMBERHOME/bin/ambpdb -p polyAT_vac.prmtop *-c*
>> polyAT_gb_init_min.rst > polyAT_gb_init_min.pdb
> so instead of a "<" I used a "-c" and it worked for me.
>
> I have a second commend: (Maybe it is a stupid questions, but I think
> it might be important for my research.)
> I would like to understand the *.prmtop files better, for example
> where can I find the definition of the periodic truncated octahedron
> (size of box, is it periodic/vacuum, which shape,...).
> Maybe there is a Manual, or an tutorial which explains the
> prmtop-Strutures.
>
> Best Regards
> Johannes Kalliauer
> TU Wien (Vienna University of Technology)
> Austria
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: summary.png)
