Date: Wed, 15 Jun 2016 10:54:35 +0800
Here. I also included python2.7/site-packages/
On Wed, Jun 15, 2016 at 10:48 AM, Hai Nguyen <nhai.qn.gmail.com> wrote:
> Please see my reply below.
>
> On Tue, Jun 14, 2016 at 10:06 PM, Abdul-Rashid Iii Sampaco <
> absampaco.up.edu.ph> wrote:
>
> > Thank you for all your responses.
> >
> > I tried Case's suggestion. I understand the command forces the computer
> to
> > download Miniconda. However, it fails to connect. The error message is:
> >
> > Connecting to repo.continuum.io (repo.continuum.io
> )|54.225.73.227|:443...
> > failed: Connection timed out
> >
> > I'm not sure if this is already a problem on my internet connection, or
> how
> > my terminal connects to the internet. I opened the link manually on the
> > browser and downloaded the shell file. I then used ./configure
> > --with-python /path/to/miniconda.sh but it cannot be run by the
> terminal. I
> > understand now that the shell file cannot be run on the terminal. Any
> > suggestions on how to run it?
> >
> > Btw, I also tried Nguyen's suggestion, but it does not work. It says it
> > cannot find the file.
> >
> >
> Can you send the output of this
>
> *ls $AMBERHOME/lib/*
>
>
> > To Chagas, I don't know exactly what you mean by "Try to copy your
> content
> > and put it in the .profile file". Which content? I'm not an experienced
> > Linux user. I'm sorry if you have to be very specific on your
> instructions
> > to me.
> >
> > Sincerely,
> >
> > Rashid
> >
> > On Tue, Jun 14, 2016 at 10:25 PM, Hai Nguyen <nhai.qn.gmail.com> wrote:
> >
> > > Hi
> > >
> > > First, do you see any folder in
> $AMBERHOME/lib/python*X.Y*/site-packages?
> > > where "X.Y" can be *2.7, 3.4, 3.5* (you should expect to see parmed,
> > > sander, pytraj, ...)
> > >
> > > If yes, do this again in your terminal
> > >
> > > source /home/absampaco/amber16//amber.sh
> > >
> > > If nothing works, please try Case's suggestion.
> > >
> > > Hai
> > >
> > > On Tue, Jun 14, 2016 at 4:22 AM, Abdul-Rashid Iii Sampaco <
> > > absampaco.up.edu.ph> wrote:
> > >
> > > > Hi,
> > > >
> > > > I am installing AmberTools16 on my laptop using Ubuntu BioLinux on
> VM.
> > I
> > > > followed all instructions on the guide. Based on what I see,
> everything
> > > > works perfectly well after make install. However, the 'make test'
> gives
> > > > this error message:
> > > >
> > > > Error: Could not import Amber Python modules! Probably your Amber
> > Python
> > > > environment was not set up correctly.
> > > >
> > > > I already added the test -f /home/absampaco/amber16//amber.sh &&
> source
> > > > /home/absampaco/amber16//amber.sh to my ~/.bash.rc before the 'make
> > > > install'.
> > > >
> > > > I would appreciate any help given.
> > > > _______________________________________________
> > > > AMBER mailing list
> > > > AMBER.ambermd.org
> > > > http://lists.ambermd.org/mailman/listinfo/amber
> > > >
> > > _______________________________________________
> > > AMBER mailing list
> > > AMBER.ambermd.org
> > > http://lists.ambermd.org/mailman/listinfo/amber
> > >
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
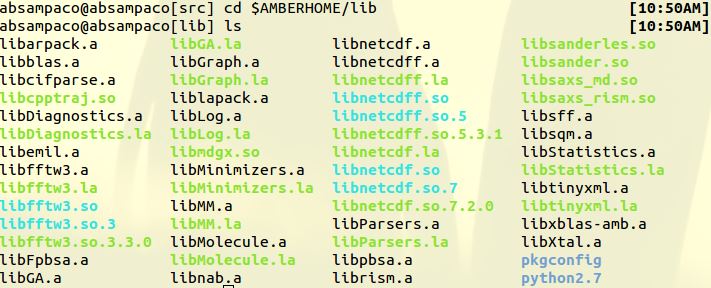
(image/jpeg attachment: lib.JPG)
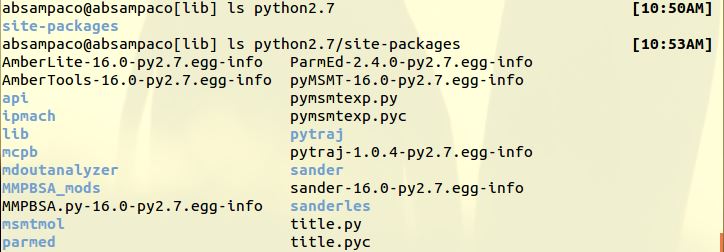
(image/jpeg attachment: site-packages.JPG)
