Date: Thu, 17 Dec 2015 22:25:51 +0000
Hello everyone, I have been modeling two small, interacting proteins and found that I can achieve significant differences in my production RMSD plots of the two proteins when I use pmemd.MPI with ig=-1. I am aware that both these settings will ensure that I have at least some differences, but these differences seem quite large.
Should all of my production runs be done in triplicate for analysis? Is this normal for production runs with my settings or is my system not reaching equilibrium yet? I searched the literature and could not find much on this topic.
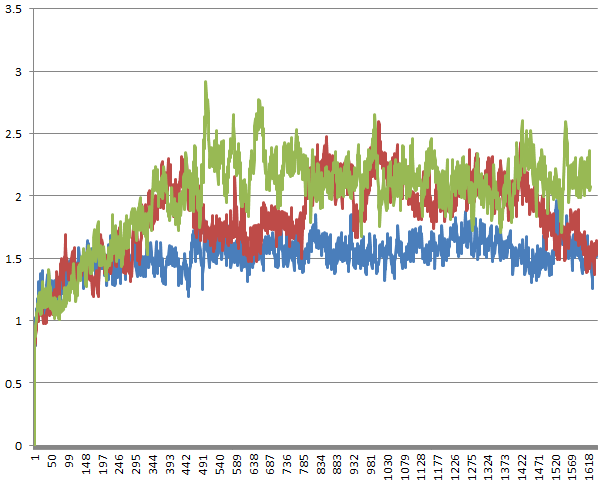
Attached is a picture of the RMSD of three identical 32ns production runs with ig=-1
John Morrow
MD Anderson Cancer Center
Graduate School of Biomedical Sciences
Experimental Therapeutics
The information contained in this e-mail message may be privileged, confidential, and/or protected from disclosure. This e-mail message may contain protected health information (PHI); dissemination of PHI should comply with applicable federal and state laws. If you are not the intended recipient, or an authorized representative of the intended recipient, any further review, disclosure, use, dissemination, distribution, or copying of this message or any attachment (or the information contained therein) is strictly prohibited. If you think that you have received this e-mail message in error, please notify the sender by return e-mail and delete all references to it and its contents from your systems.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: 3runs.backboneRMSD_32ns.png)
