Date: Thu, 10 Dec 2015 17:05:43 +0100
Dear Pengfei and Dear Carlos and dear AMBER
Good evening...
I have managed to follow ZAFF.prep and ZAFF.frcmod (AMBER) way to handle
the zinc coordination.
With non-bonded model (different way to handle zinc) there was a problem
with CYS CYS bond formation during simulated annealing.
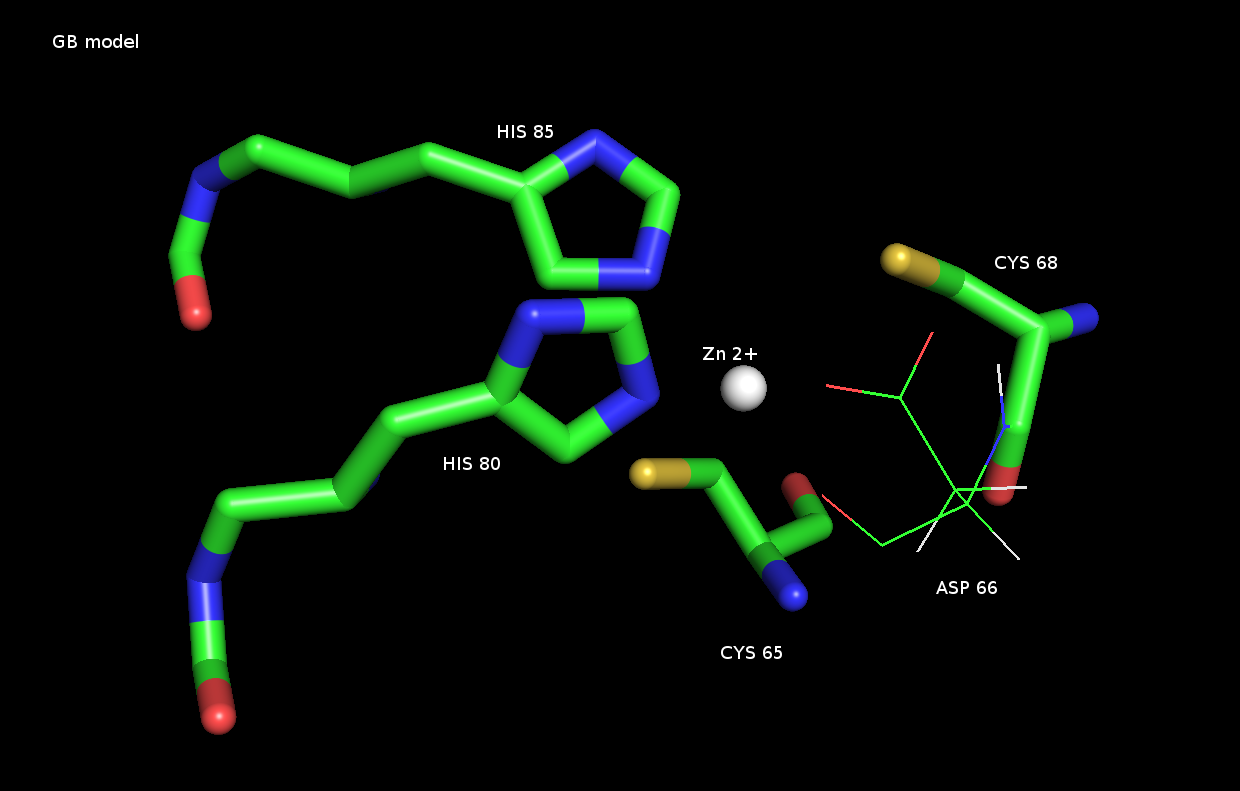
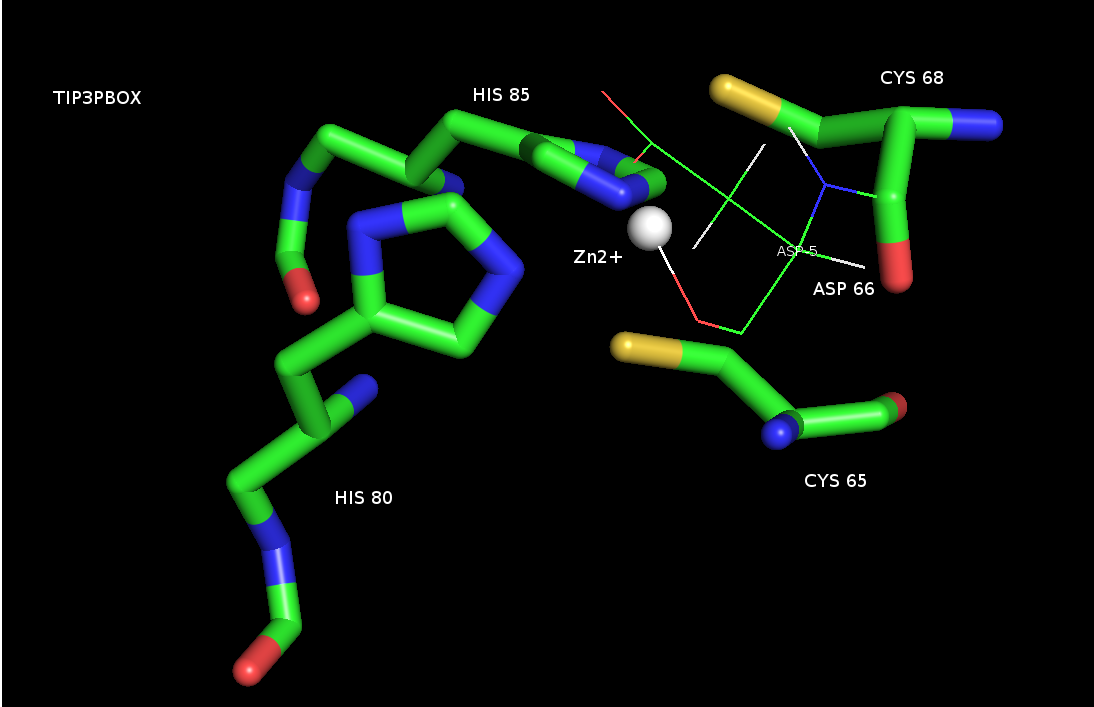
I followed ZAFF.prep and ZAFF.frcmod (AMBER) as suggested by Pengfeim now
my coordination sphere is ok. But I have another problem, zinc atom is
forming
a bond with the side chain of the aspartic acid. I have neutralised my
system to net charge 0 in TIP3. Even I observed same problem in GB model
but when I extract
low energy structures there zinc-ASP side chain bond was absent.
Is this such behaviour of Zn2+ is common even after defined a 2SG-CYS and
2-NE2-HIS bond during prmrop and inpcrd files generation ? I have attached
the image.
I would highly appreciate your suggestions and ideas...
thanks in advance ...
Best wishes
VINCE
PS: simulated annealing + NMR restraints on.
[image: Inline images 4][image: Inline images 3]
On 1 December 2015 at 08:52, V. Kumar <vin.vasanth.gmail.com> wrote:
> Dear Pengfei
>
> Good morning.
>
> thank you so much for your response. I will to follow tutorial link.
>
> Have a nice day..
>
> thanks a lot
> Vince
>
> On 30 November 2015 at 22:58, Carlos Simmerling <
> carlos.simmerling.gmail.com> wrote:
>
>> I don't know exactly what's going on, but if it shows the S-S bond from
>> the
>> prmtop and not the bonds to the Zn, then something went wrong. I think you
>> need to read the leap output carefully, not just the "OK". read what you
>> asked it to do and any information that it wrote in response. the bonds do
>> not seem right.
>>
>> On Mon, Nov 30, 2015 at 3:38 PM, V. Kumar <vin.vasanth.gmail.com> wrote:
>>
>> > Dear Carlos
>> >
>> > Thanks a lot for your response.
>> >
>> > Yes I have used prmtop and inpcrd to generate that image.
>> >
>> > I followed the following procedure.
>> >
>> > First I have created a linear structure using tleap. Next I have include
>> > the zinc metal ion information as a residue. Like Zn(atom) in
>> ZNA(residue).
>> > I took my peptide with zna residue coordinates then I bonded Zn to 2CYS
>> and
>> > 2HIS, to save prmtop and inpcrd was not successful then I will input
>> > zinc.frcmod (the file in my first email). Then I could easily save my
>> > parmtop and inpcrd. Then I have also checked the unit in tleap, I got
>> "OK".
>> >
>> > I actually modified force field to handle zinc on my own, I found those
>> > values on the internet.
>> >
>> > I have followed CU+ | Plastocyanin tutorial
>> > http://ambermd.org/tutorials/advanced/tutorial1_orig/section2.htm.
>> >
>> > In addition my HIS are nonstandard...
>> >
>> > As per the tutorial my procedure to generate prmtop and inpcrd with zn
>> > metal ion information is correct and I m not sure about the parameter
>> file.
>> >
>> > thanks you so much...
>> >
>> > Vince
>> >
>> >
>> > On 30 November 2015 at 18:02, Carlos Simmerling <
>> > carlos.simmerling.gmail.com
>> > > wrote:
>> >
>> > > how did you generate that picture? did it use a prmtop file for the
>> > bonds,
>> > > or distances?
>> > > are you sure that the parameters you list were used to generate your
>> > > prmtop? it doesn't seem right - you need to look carefully at the leap
>> > > outfile file.
>> > >
>> > > On Mon, Nov 30, 2015 at 12:00 PM, V. Kumar <vin.vasanth.gmail.com>
>> > wrote:
>> > >
>> > > > Dear AMBER
>> > > >
>> > > > I have successfully complete refinement of my peptide with NMR
>> > restraints
>> > > > on. For zinc coordination I have used following parameter (bellow).
>> In
>> > > the
>> > > > end of calculation I lost perfe tetrahedral symmetry. Two cyestein
>> > > residues
>> > > > are forming covalent bond with each other in water or
>> > C65--SH<===>SH(C68)
>> > > > about 1.5A (I have attached the image). Could any body suggest me
>> why I
>> > > > couldn't maintain proper tetrahedral geometry and why cysteins
>> forming
>> > > > covalent bonds. I am using non bonded zinc model.
>> > > >
>> > > >
>> > > >
>> > > >
>> > > > thanks you in advance...
>> > > > Vince
>> > > >
>> > > >
>> > > > # modifications to force field for zinc finger domain
>> > > >
>> > > > my zinc coordination frcmod file...
>> > > > MASS
>> > > > ZN 65.36
>> > > >
>> > > > BOND
>> > > > NB-ZN 70.000 2.05000 #kludge by JRS
>> > > > ZN-S 70.000 2.10000 #kludge by JRS
>> > > > ZN-SH 70.000 2.90000 #for pcy
>> > > > CT-SH 222.000 1.81000 #met(aa)
>> > > >
>> > > > ANGLE
>> > > > ZN-NB-CV 50.000 126.700 #JRS estimate
>> > > > ZN-NB-CR 50.000 126.700 #JRS estimate
>> > > > ZN-NB-CP 50.000 126.700 #JRS estimate
>> > > > ZN-NB-CC 50.000 126.700 #JRS estimate
>> > > > ZN-SH-CT 50.000 120.000 #JRS estimate
>> > > > ZN-S -CT 50.000 120.000 #JRS estimate
>> > > > ZN-S -C2 50.000 120.000 #JRS estimate
>> > > > ZN-S -C3 50.000 120.000 #JRS estimate
>> > > > NB-ZN-NB 10.000 110.000 #dac estimate
>> > > > NB-ZN-SH 10.000 110.000 #dac estimate
>> > > > NB-ZN-S 10.000 110.000 #dac estimate
>> > > > SH-ZN-S 10.000 110.000 #dac estimate
>> > > > ZN-SH-CT 50.000 120.000 #JRS estimate
>> > > > CT-CT-SH 50.000 114.700 #met(aa)
>> > > > HC-CT-SH 35.000 109.500
>> > > > H1-CT-SH 35.000 109.500
>> > > > CT-SH-CT 62.000 98.900 #MET(OL)
>> > > > SH-ZN-SH 10.00 113.000 #by VK
>> > > > S-ZN-S 10.00 109.100 #by VK
>> > > >
>> > > > DIHE
>> > > > X -NB-ZN-X 1 0.000 180.000 3.000
>> > > > X -ZN-SH-X 1 0.000 180.000 3.000
>> > > > X -ZN-S -X 1 0.000 180.000 3.000
>> > > > X -CT-SH-X 3 1.000 0.000 3.000
>> > > >
>> > > >
>> > > > NONBON
>> > > > ZN 1.10 0.0125
>> > > >
>> > > > _______________________________________________
>> > > > AMBER mailing list
>> > > > AMBER.ambermd.org
>> > > > http://lists.ambermd.org/mailman/listinfo/amber
>> > > >
>> > > >
>> > > _______________________________________________
>> > > AMBER mailing list
>> > > AMBER.ambermd.org
>> > > http://lists.ambermd.org/mailman/listinfo/amber
>> > >
>> > _______________________________________________
>> > AMBER mailing list
>> > AMBER.ambermd.org
>> > http://lists.ambermd.org/mailman/listinfo/amber
>> >
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>>
>
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: zinc_cordination2.png)
(image/png attachment: TIP3_ZN.png)
