Date: Fri, 2 Oct 2015 13:54:07 -0400
Dear AMBER experts,
A student of mine is having an interesting problem come up that I do not
understand. We are modeling salt crystals and have done extensive
convergence testing for all of the simulations we are running. We have
sorted out everything except one issue. We are building salt crystals and
evaluating their energies (no dynamics) using periodic boundary
conditions. We have run calculations using systems of various sizes. In
our case the smallest unit is 8 ions: 4 Na+ and 4 Cl- forming a small
cube. We have built systems that are 5x5x5, 10x10x10, and so on of these
tiny units. It should be the case that the system energy on a per ion
basis is exactly the same for all of these different systems. However,
this is not quite the case and it turns out that the VDW energy is the
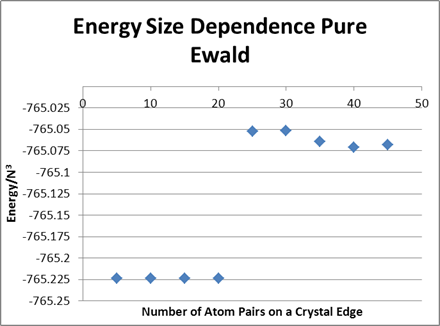
source of the inconsistency. The VDW energy is the same out to 8 or 9
digits for systems with 5, 10, 15, and 20 units (per edge), but then there
is a significant jump (0.3%) in energy between 20 and 25 units. I've
inserted a table and a plot below that illustrate this. Note that the
table has total energy, electrostatic, and vdw parsed out. The figure has
just the total energy. And note that in the table we divided by (2N)^3 so
it really is the energy per particle, whereas we just divided by N^3 in the
figure; don't be distracted by the factor of 8.
We have double-checked that the mdin files are identical, so I don't think
it is the case that we have made a silly mistake. Does this dependence of
the system VDW energy on the system size ring any bells for anyone? For
now we are assuming that the 5--20 data are 'correct' and that there is
something wrong with the others, but if anyone has any insight to this, I
would love to hear it.
Thanks,
Brent
N Atom pairs per crystal edge Energy (kcal/mol) Energy/(2N)3 Eelec Eelec/
(2N)3 Evdw Evdw/(2N)3 5 -95652.9106 -95.6529106 -102703.1589 -102.7031589
7050.2482 7.0502482 10 -765223.2851 -95.65291064 -821625.2708 -102.7031589
56401.9857 7.050248213 15 -2582628.587 -95.65291064 -2772985.289
-102.7031589 190356.7016 7.050248207 20 -6121786.281 -95.65291064
-6573002.167 -102.7031589 451215.8854 7.050248209 25 -11953946.43
-95.63157141 -12837894.86 -102.7031589 883948.4309 7.071587447 30
-20656397.79 -95.63147123 -22183882.31 -102.7031588 1527484.525 7.071687614
35 -32802143.74 -95.63307213 -35227183.48 -102.7031588 2425039.741
7.070086709 40 -48964521.92 -95.63383187 -52584017.33 -102.7031589
3619495.414 7.06932698 45 -69716793.27 -95.63346127 -74870602.81
-102.7031589 5153809.542 7.069697588
[image: Inline image 2]
-- _______________________________________________ Brent P. Krueger.....................phone: 616 395 7629 Professor.................................fax: 616 395 7118 Hope College..........................Schaap Hall 2120 Department of Chemistry Holland, MI 49423
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
