Date: Sun, 9 Aug 2015 08:51:02 +0200
Dear AMBER users,
Wish you are fine
I performed a 30 ns MD simulation in 3 stages, using AMBER, each stage for
10 ns.
*Description of the simulation:*
I used this md control file for all stages
Production
&cntrl
imin=0,
ntx=5,
irest=1,
nstlim=5000000,
dt=0.002,
ntf=2,
ntc=2,
temp0=310.0,
tempi=310.0,
ntpr=5000,
ntwx=5000,
cut=8.0,
ntb=2,
ntp=1,
ntt=3,
gamma_ln=1.0,
/
*The question is**:*
Reagarding the last line in the following cpptraj input,
> trajin mol500001.crd #From stage 1
> trajin mol500002.crd #From stage 2
> trajin mol500003.crd #From stage 3
> reference mol5min.res
> autoimage
> rms reference mass out 02_03.rms time 10 :44-61
>
1- Does the " time 10 " indicate the time of each frame in ps ?
This how I calculated the value of 10
No. of frames = nstlim/ ntpr =5000000/5000=1000 frame
Simulation time =nstlim*dt = 5000000*0.002=10000 ps
Time of each frame = simulation time/no.frames= 10 ps
And
2- Does " :44-61 " mean to specify residues from 44-61 only?
Should I also add the command " strip: WAT " to exclude all water molecules
from RMSD ?
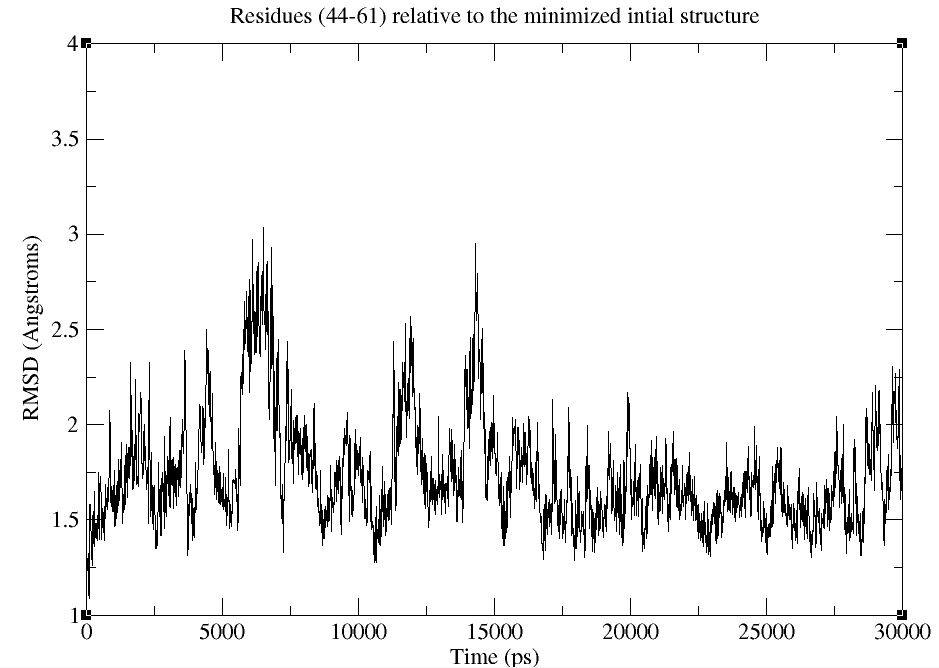
This is the graph that I got
So does this graph represent the time of MD simulation in (ps) on x-axis
and the RMSD in (Angstroms) on y-axis ?
I just want to double check my work because I am not sure that I well
understand the part concerned with cpptraj in AmberTools12 Manual
This is also the Cpptraj_logfile
CPPTRAJ: Trajectory Analysis. V13.0
___ ___ ___ ___
| \/ | \/ | \/ |
_|_/\_|_/\_|_/\_|_
AmberParm Title: [default_name]
Radius Set: modified Bondi radii (mbondi)
INPUT: Reading Input from file rmsd.cpptraj
[trajin mol500001.crd]
[mol500001.crd] contains 1000 frames.
[trajin mol500002.crd]
[mol500002.crd] contains 1000 frames.
[trajin mol500003.crd]
[mol500003.crd] contains 1000 frames.
[reference mol5min.res]
[mol5min.res] contains 1 frames.
[mol5min.res] is an AMBER restart file, no velocities, Parm mol5.top
(Orthogonal box) (reading 1 of 1)
[autoimage]
AUTOIMAGE: To box center based on center of mass, anchor is first
molecule.
[rms reference mass out 02_03.rms time 10 :44-61]
RMSD: (:44-61), reference is reference frame mol5min.res (:44-61), with
fitting, mass-weighted.
PARAMETER FILES:
0: mol5.top, 23856 atoms, 7205 res, box: Orthogonal, 7033 mol, 7024
solvent, 3000 frames
INPUT TRAJECTORIES:
0: [mol500001.crd] is an AMBER trajectory, Parm mol5.top (Orthogonal box)
(reading 1000 of 1000)
1: [mol500002.crd] is an AMBER trajectory, Parm mol5.top (Orthogonal box)
(reading 1000 of 1000)
2: [mol500003.crd] is an AMBER trajectory, Parm mol5.top (Orthogonal box)
(reading 1000 of 1000)
Coordinate processing will occur on 3000 frames.
REFERENCE COORDS:
The following 1 frames have been defined:
0: mol5min.res frame 1
Active reference frame for masks is 0
OUTPUT TRAJECTORIES:
No files.
BEGIN TRAJECTORY PROCESSING:
.....................................................
ACTION SETUP FOR PARM [mol5.top] (2 actions):
0: [autoimage]
Anchor molecule is 1
7032 molecules are mobile.
1: [rms reference mass out 02_03.rms time 10 :44-61]
Mask [:44-61] corresponds to 272 atoms.
----- [mol500001.crd] (1-1000, 1) -----
0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Complete.
----- [mol500002.crd] (1-1000, 1) -----
0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Complete.
----- [mol500003.crd] (1-1000, 1) -----
0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Complete.
Read 3000 frames and processed 3000 frames.
ACTION OUTPUT:
DATASETS:
1 data set:
RMSD_00000 "RMSD_00000" (double), size is 3000
DATAFILE OUTPUT:
02_03.rms: RMSD_00000
02_03.rms: Writing 3000 frames.
Thanks in advance!
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: 44-61.png)
