Date: Fri, 17 Apr 2015 00:51:08 -0600
Dear users,
I am trying to simulate a carbon nanotube in water. There are lot's of
papers, so I have the parameters for each field of .frcmod file. I have
also generated prmtop and inpcrd file from leap, where I charged ff14SB,
gaff and cnt.frcmod (from parmchk2 and then edited to correct some
parameters to write papers found and accepted parameters).
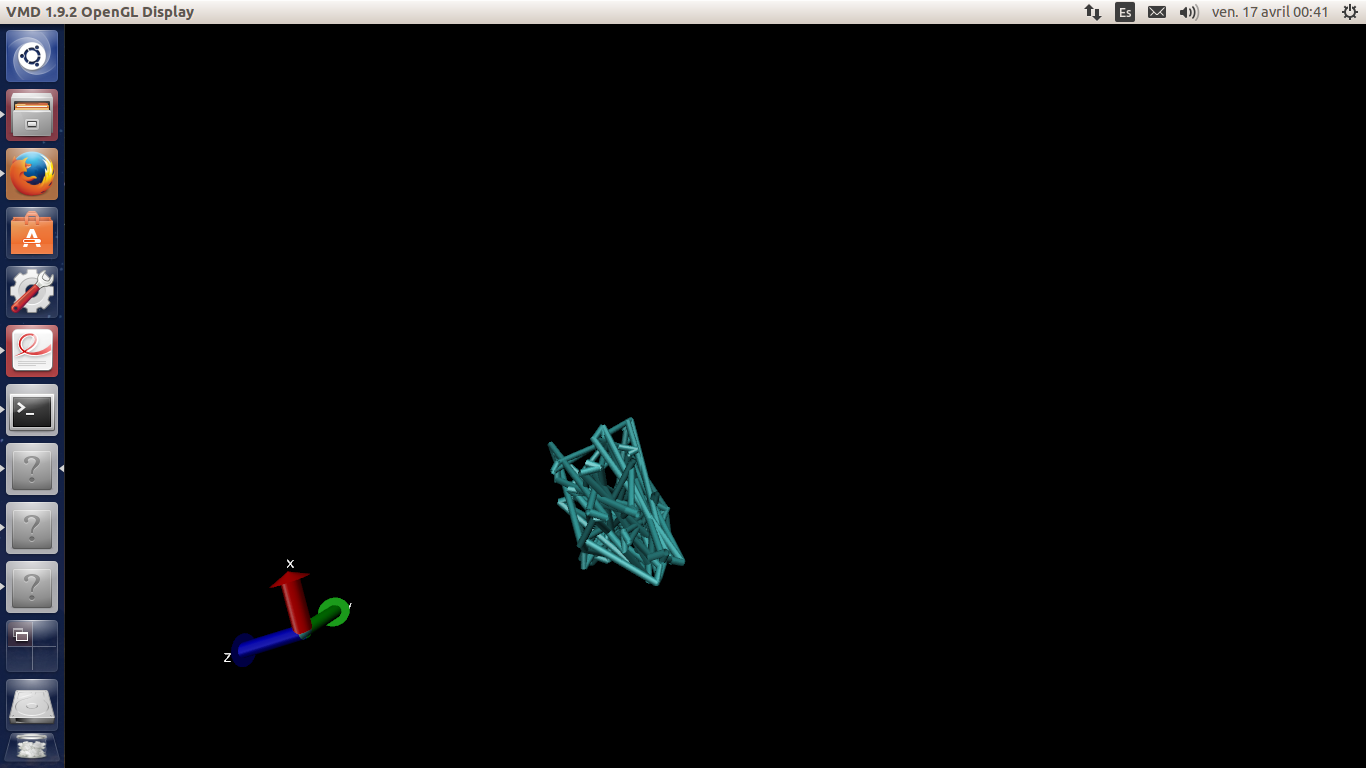
*The problem* : After the minimisation and heating (300K), when I visualize
heat.mdcrd file all carbon atoms are missplaced in the box extremities and
waters molecules take the carbon nanotube place... The carbon nanotube is
deaaad!
This kind of behavior is revelating a wrong force field, right? Do I have
to edit a .lib file and charge it with loadOff in leap for the carbon
nanotube molecule?
Thank you a lot for your time and possible answers o ways to solve this
problem!
*Pierre BERTIN*
Bioinformatics and Biostatistics student (MSc),
University Paris-Sud XI, ORSAY.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: CNT_endsimul.png)
