Date: Wed, 25 Feb 2015 17:57:43 +0100
Dear Amber Developers,
I am trying to run a simulation of H-REMD on a membrane protein.
I am using Amber14 with the ff14SB and gaff force fields.
My system comprizes the channel protein, a small patch of membrane
and water and ions on both sides. My aim is to bias the backbone
dihedrals of the N-terminal tail of the protein rescaling the force
costant by factors 1.0, 0.9, 0.8, 0.7, 0,6, 0.5, 0.4, 0.3. The
topology files have been edited using parmed.py. For instance to
rescale the dihedral force constants by a factor of 0.5 I used the
script:
#Backbone phi angles
deleteDihedral :1-36.C :2-37.N :2-37.CA :2-37.C
addDihedral :1-36.C :2-37.N :2-37.CA :2-37.C 0.0000 4.0000
0.0000 1.2000 2.0000 type multiterm
addDihedral :1-36.C :2-37.N :2-37.CA :2-37.C 0.0000 1.0000
0.0000 1.2000 2.0000 type multiterm
addDihedral :1-36.C :2-37.N :2-37.CA :2-37.C 0.1350 2.0000
0.0000 1.2000 2.0000 type multiterm
addDihedral :1-36.C :2-37.N :2-37.CA :2-37.C 0.2100 3.0000
0.0000 1.2000 2.0000
#Backbone psi angles
deleteDihedral :2-36.N :2-36.CA :2-36.C :3-37.N
addDihedral :2-36.N :2-36.CA :2-36.C :3-37.N 0.2250 1.0000
180.0001 1.2000 2.0000 type multiterm
addDihedral :2-36.N :2-36.CA :2-36.C :3-37.N 0.7900 2.0000
180.0001 1.2000 2.0000 type multiterm
addDihedral :2-36.N :2-36.CA :2-36.C :3-37.N 0.2750 3.0000
180.0001 1.2000 2.0000 type multiterm
addDihedral :2-36.N :2-36.CA :2-36.C :3-37.N 0.0000 4.0000
0.0000 1.2000 2.0000
parmout vdac2_trim_0.5.prmtop
Each of the eight replicas was minimized using its own topology file:
&cntrl
imin = 1,
maxcyc = 2000,
ncyc = 1000,
ntb = 1,
ntr = 0,
cut = 8.0,
ntwr = 2000,
ntpr = 100,
/
sander -O -i min.in -o min.out -p vdac2_trim_1.0.prmtop -c
hvdac2-ions1M_trim.inpcrd -r vdac2_trim_min_1.0.rst
After the minimization each replica was equilibrated in the NPT ensemble
using its own topology file:
Equilibration
&cntrl
irest=0, ntx=1,
nstlim=2500000, dt=0.002,
ntt=3, gamma_ln=1.0,
temp0=300.0,
ntc=2, ntf=2, nscm=1000,
ntb=2, ntp=2, cut=8.0,
ntpr=100, ntwx=100, ntwr=1000,
/
Up to this point everything was fine and I did not experience any kind of
problem. Finally I tried to run the H-REMD simulation using as input
configurations the restart structures yielded by the equilibration. My
input files (indeed all equal) are as follows:
H-REMD
&cntrl
irest=0, ntx=1,
nstlim=1250, dt=0.002,
ntt=3, gamma_ln=1.0,
tempi=300.0, temp0=300.0,
ig=-1, iwrap=1,
ntc=2, ntf=2, nscm=1000,
ntp=0, ntb=1, cut=8.0,
ntxo=2, ioutfm=1,
ntpr=1250, ntwx=1250, ntwr=1250,
numexchg=100,
/
I used the following groupfile:
-O -rem 3 -remlog rem_R1.log -i hremd.mdin0.001 -o hremd.mdout_R1.001 -c
vdac2_trim_equil.rst_R2.001 -r hremd.rst_R1.001 -x hremd.netcdf_R1.001
-inf hremd.mdinfo_R1.001 -p vdac2_trim_1.0.prmtop
-O -rem 3 -remlog rem_R1.log -i hremd.mdin0.002 -o hremd.mdout_R1.002 -c
vdac2_trim_equil.rst_R2.002 -r hremd.rst_R1.002 -x hremd.netcdf_R1.002
-inf hremd.mdinfo_R1.002 -p vdac2_trim_0.9.prmtop
-O -rem 3 -remlog rem_R1.log -i hremd.mdin0.003 -o hremd.mdout_R1.003 -c
vdac2_trim_equil.rst_R2.003 -r hremd.rst_R1.003 -x hremd.netcdf_R1.003
-inf hremd.mdinfo_R1.003 -p vdac2_trim_0.8.prmtop
-O -rem 3 -remlog rem_R1.log -i hremd.mdin0.004 -o hremd.mdout_R1.004 -c
vdac2_trim_equil.rst_R2.004 -r hremd.rst_R1.004 -x hremd.netcdf_R1.004
-inf hremd.mdinfo_R1.004 -p vdac2_trim_0.7.prmtop
-O -rem 3 -remlog rem_R1.log -i hremd.mdin0.005 -o hremd.mdout_R1.005 -c
vdac2_trim_equil.rst_R2.005 -r hremd.rst_R1.005 -x hremd.netcdf_R1.005
-inf hremd.mdinfo_R1.005 -p vdac2_trim_0.6.prmtop
-O -rem 3 -remlog rem_R1.log -i hremd.mdin0.006 -o hremd.mdout_R1.006 -c
vdac2_trim_equil.rst_R2.006 -r hremd.rst_R1.006 -x hremd.netcdf_R1.006
-inf hremd.mdinfo_R1.006 -p vdac2_trim_0.5.prmtop
-O -rem 3 -remlog rem_R1.log -i hremd.mdin0.007 -o hremd.mdout_R1.007 -c
vdac2_trim_equil.rst_R2.007 -r hremd.rst_R1.007 -x hremd.netcdf_R1.007
-inf hremd.mdinfo_R1.007 -p vdac2_trim_0.4.prmtop
-O -rem 3 -remlog rem_R1.log -i hremd.mdin0.008 -o hremd.mdout_R1.008 -c
vdac2_trim_equil.rst_R2.008 -r hremd.rst_R1.008 -x hremd.netcdf_R1.008
-inf hremd.mdinfo_R1.008 -p vdac2_trim_0.3.prmtop
Finally, I tried to run the job using the GPUs:
#!/bin/bash
source /etc/modules.sh
module load intel openmpi amber14
export CUDA_VISIBLE_DEVICES=0,1,2,3
mpirun -np 8 pmemd.cuda.MPI -ng 8 -groupfile hremd.groupfile
The job unfortunately aborted almost immediately yielding
nan and anomalous energy values from the very first steps.
I also received the error message:
Error: unspecified launch failure launching kernel kClearForces
cudaFree GpuBuffer::Deallocate failed unspecified launch failure
I verified that:
1) The input configurations are not corrupt. In fact, if I restart
the equilibration run, this proceeds without problems.
2) The topology files also should be correct, otherwise I should
have had problems during the equilibration.
3) The protocol and input files for H-REMD are the same I used in
an H-REMD simulation of an other system (the N-tail peptides of
the channel protein in an octahedral water box) that didn't
cause any problem.
When I tried to re-run the simulation on the CPUs,
mpirun -np 8 sander.MPI -ng 8 -groupfile hremd.groupfile
the simulation did not abort. However in the mdout file I received the
following error message that was repeated at every printout of the
energies:
NetCDF error: NetCDF: Variable not found
at write temp0
Moreover, the exchanges between replicas were never accepted, because
apparently the energy difference between replicas was too large. Here
is a sample of my rem.log file:
# Rep#, Neibr#, Temp0, PotE(x_1), PotE(x_2), left_fe, right_fe, Success,
Success rate (i,i+1)
# exchange 1
1 8 300.00 -56959.55********** -Infinity 0.00 F
0.00
2 3 300.00 -57029.36********** 0.00 -Infinity F
0.00
3 2 300.00 -57067.42********** -Infinity 0.00 F
0.00
4 5 300.00 -57216.37********** 0.00 -Infinity F
0.00
5 4 300.00 -57203.80********** -Infinity 0.00 F
0.00
6 7 300.00 -57135.97********** 0.00 -Infinity F
0.00
7 6 300.00 -57065.04********** -Infinity 0.00 F
0.00
8 1 300.00 -57193.50********** 0.00 -Infinity F
0.00
# exchange 8
1 2 300.00 -56742.97********** -Infinity -Infinity F
0.00
2 1 300.00 -57190.23********** -Infinity -Infinity F
0.00
3 4 300.00 -57033.87********** -Infinity -Infinity F
0.00
4 3 300.00 -57127.098096676.91 -Infinity -Infinity F
0.00
5 6 300.00 -57154.20********** -Infinity -Infinity F
0.00
6 5 300.00 -57068.91********** -Infinity -Infinity F
0.00
7 8 300.00 -57037.624099387.93 -Infinity -Infinity F
0.00
8 7 300.00 -57033.40********** -Infinity -Infinity F
0.00
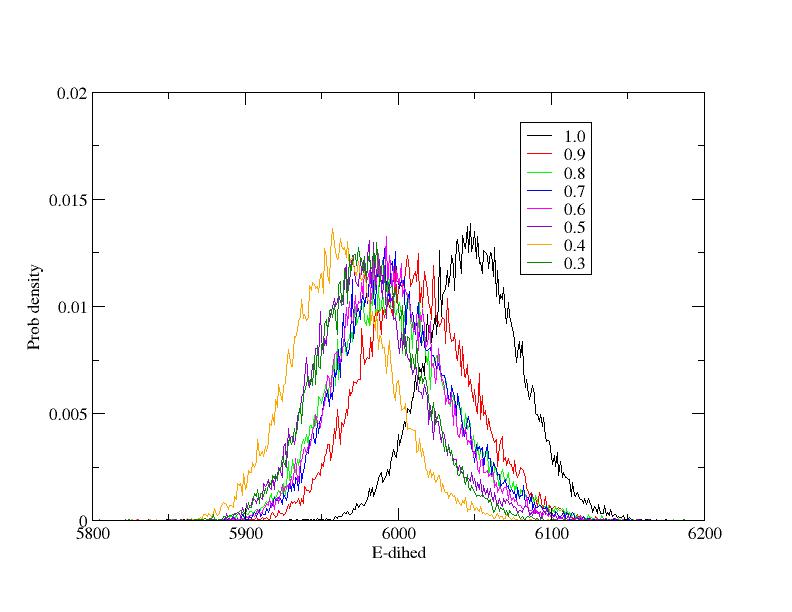
Indeed the energy distributions at the end of the equilibration are not
wonderful since some of the distributions are probably too much
overlapping (see attached Figure). In such a case however, the problem
should be the opposite as the one I observed: the replicas should exchange
too often. In any case I checked that the dihedral force constants in the
topology files correspond to the values I set with parmed.py.
In the hypothesis that this could be a problem of precision, I asked our
system administrator to re-compile the Amber14 code using DPFP. In this
case however I got the error message: "unknown flag: -ng" which means that
the code in double precision is uncapable of running parallel jobs with
multisander.
Does anyone imagine where this problem comes from ? How could I fix it ?
Any suggestion will be
more than welcome.
Many thanks for the help and kind regards,
Carlo Guardiani
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: Edihed_Equil.jpg)
