Date: Sat, 7 Feb 2015 23:22:43 +0200
Dear Sir
wish you are fine !
I am running a md simulation on protein in a water box of TIP3P water model
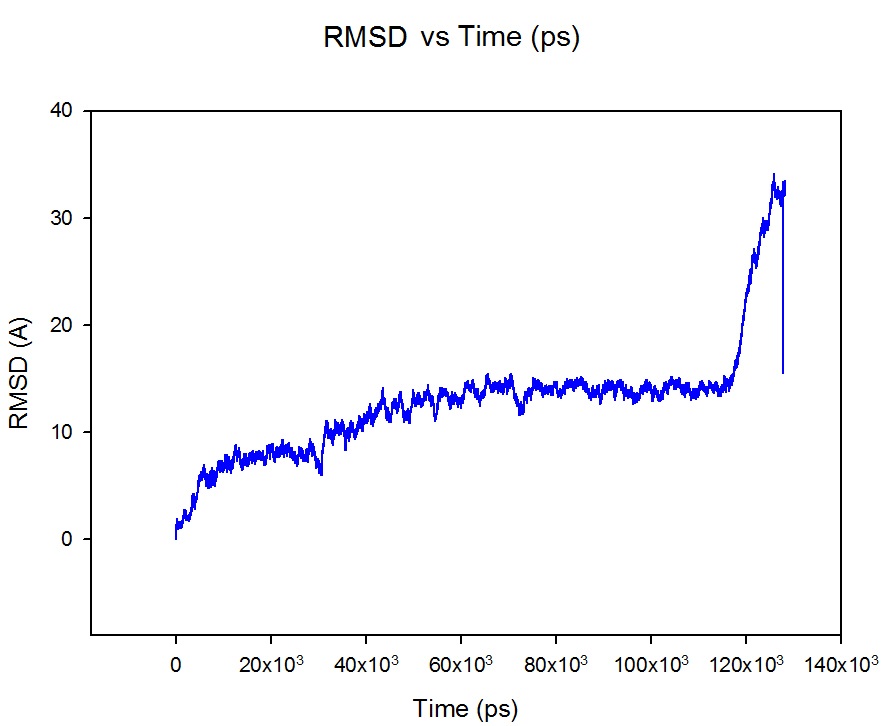
under ff99 force field . After 117 ns of simulation I thought that the
protein has reached the equilibrium, However it starts to partially unfold
after that . That was very obvious and illogical to me because the protein
tolerated 117 ns of simulation with some favorable and reasonable local
conformational change and after that starts to unfold. the used protocol is
included below. Also, The RMSD graph is attached !
What do you think the reason of this behavior?
Do you think the old force field of ff99 may be the reason for this ?
Do think it is an accidental behavior that may not be repeated if I
re-start the simulation from good earlier restart file?
what do you think may be the source of errors in this protocol ?!
1- 800 steps of minimization round 1 with restrains
> > &cntrl
> > imin=1, nmropt=0,
> > ntx=1, irest=0,
> > ntxo=1, ntpr=100, ntrx=1, ntwr=100, ntwx=100,
> > ntf=2, ntb=1, igb=0, ntc=2, cut=10.0,
> > maxcyc=800, ncyc=100, ntmin=1,
> > ibelly=0, ntr=1, restraint_wt=5.0, restraintmask=':1-175',
> > &end
>
> >
> > 2- heating up to 100 K
> > &cntrl
> > imin=0, nmropt=0,
> > ntx=1, irest=0,
> > ntxo=1, ntpr=100, ntrx=1, ntwr=100, ntwx=100,
> > ntf=2, ntb=2, igb=0, ntc=2, cut=10.0,
> > nstlim=50000, nscm=500, nrespa=1, dt=0.001,
> > Tempi=0.0, Temp0=100.0, ntt=3, gamma_ln=2.0,
> > pres0=1.0, ntp=1, taup=1.0,
> > ntr=1, restraint_wt=5.0, restraintmask=':1-175',
> > &end
>
> >
> > 3- 800 steps of minimization round 2 without restrains
> > &cntrl
> > imin=1, nmropt=0,
> > ntx=1, irest=0,
> > ntxo=1, ntpr=100, ntrx=1, ntwr=100, ntwx=100,
> > ntf=2, ntb=1, igb=0, ntc=2, cut=10.0,
> > maxcyc=800, ncyc=100, ntmin=1,
> > &end
> >
>
> > 4- heating up to 310 (over 9 rounds each of 50 ps and heating upto 25 K
> > except the last step) with restrains
> > &cntrl
> > imin=0, nmropt=0,
> > ntx=1, irest=0,
> > ntxo=1, ntpr=100, ntrx=1, ntwr=100, ntwx=100,
> > ntf=2, ntb=2, igb=0, ntc=2, cut=10.0,
> > nstlim=50000, nscm=500, nrespa=1, dt=0.001,
> > Tempi=100.0 , Temp0=125.0 , ntt=3, gamma_ln=2,
> > pres0=1.0, ntp=1, taup=0.05,
> > ntr=1, restraint_wt=5.0, restraintmask=':1-175',
> > &end
>
> > 5- production MD
> > &cntrl
> > imin=0, nmropt=0,
> > ntx=5, irest=1, ntrx=1,
> > ntxo=1, ntpr=500, ntwr=500, iwrap=1, ntwx=500,
> > ntf=2, ntb=2, igb=0, ntc=2, cut=10.0,
> > nstlim=50000, nscm=500, nrespa=1, dt=0.002,
> > Tempi=310.0, Temp0=310.0, ntt=3, gamma_ln=2,
> > pres0=1.0, ntp=1, taup=2.0,
> > &end
Best Regards
Thanks in advance !
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: 128ns-RMSD_175_vs_Time.JPG)
