Date: Thu, 13 Nov 2014 14:43:55 +0800 (GMT+08:00)
-----原始邮件-----
发件人: "王漠野" <wangmoye13.mails.ucas.ac.cn>
发送时间: 2014年11月3日 星期一
收件人: amber <amber.ambermd.org>
抄送:
主题: The question about REMD exchange ratio
Dear AMBER users,
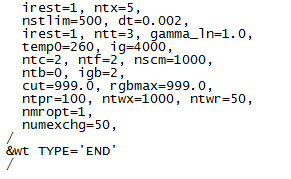
I had a REMD process with amber. One simulation time is not enough and I restarted the simulation of second times . I fixed the **.mdin files. Reset the parameters irest=1, ntx=5 and the groupfile commands were as follows:
-A -rem 1 -remlog rem2.log -i remd.mdin.00X -o remd2.mdout.00X -c remd2.rst.00X -r remd2.rst.00X -x remd2.mdcrd.00X -inf remd2.mdinfo.00X -p protein.prmtop
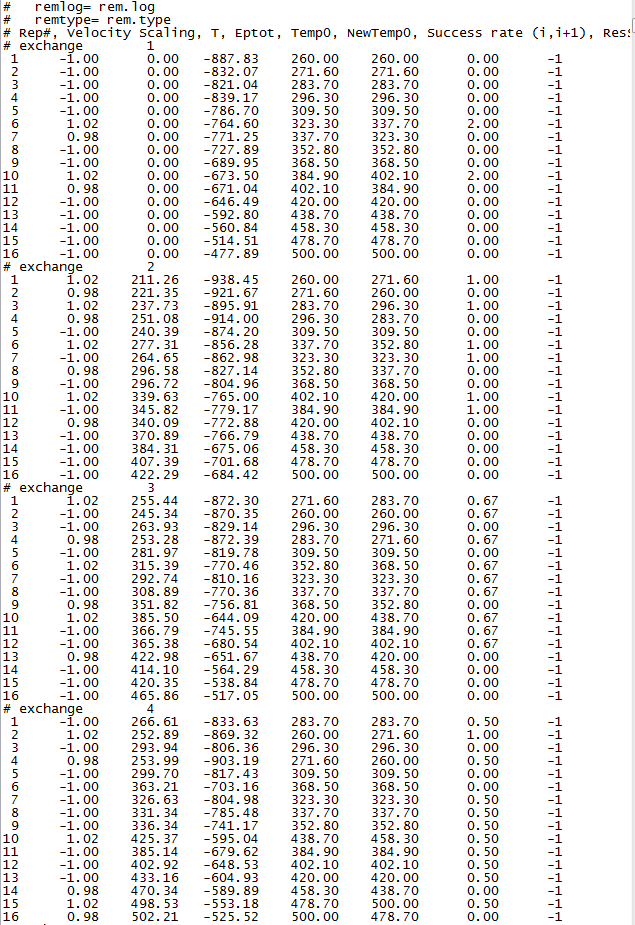
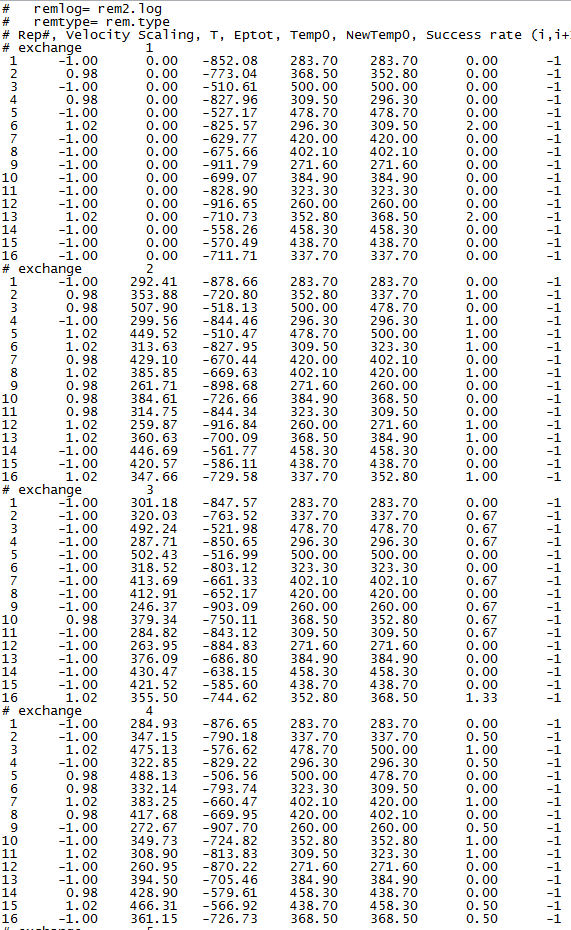
The simulation ended normally. Then I checked the rem.log file and the rem2.log file. Then I found that the exchange ratios was inconsecutive. The followed picture was the screen capture of my rem.log file and my rem2.log file:
It is a normal performance that the exchange ratios were changed from 0? Did I do something wrong or set the parameters improperly? Here I also paste my **.mdin parameters.
Is there anyone who knows the mistakes? Or the exchange ratios in amber restart REMD simulations are just how it is like this? Please let me know. Thank you !
Wang moye
University of Chinese Academy of Sciences
-- 王漠野
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: 1.png)
(image/png attachment: 2.png)
(image/png attachment: QQ______20141103200906.png)
