Date: Tue, 4 Nov 2014 10:07:39 +0000
Hi,
I am currently running a few tests with softcore TI in pmemd and I may
have found a bug when SHAKE is applied to the TI region.
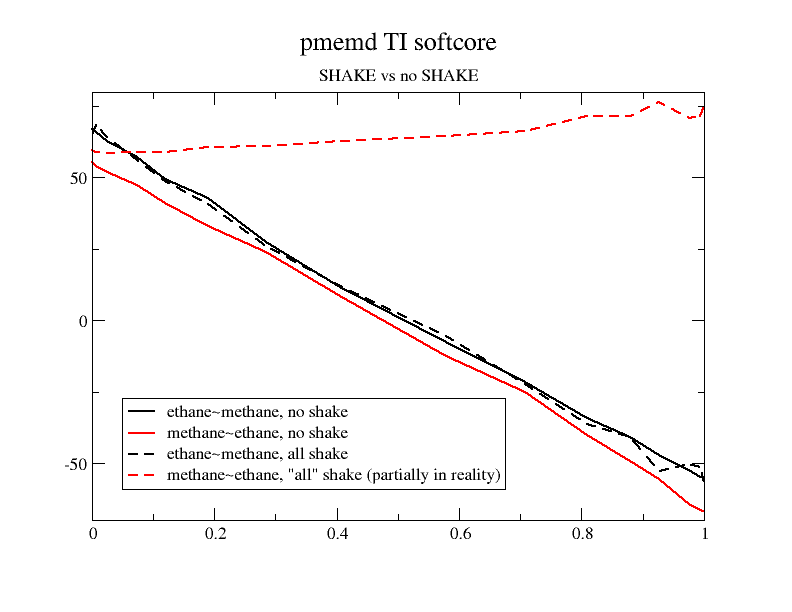
One of my test cases is ethane to methane (single-leg). To check I
have also swapped the order of ligands in the parmtop. When I switch
off SHAKE for all atoms with noshakemask both simulation series result
in virtually the same free energy gradient profile but with all SHAKE
on I get a wildly different result in the case of the methane to ethane
transformation. The reason is apparently that the softcore atoms
(three methyl hydrogens) are not SHAKEn while they are in the "reverse"
transformation. In fact, I had to reduce the time step to 0.001 to be
able to run it. General question: can I actually expect the no shake
profile to track the all shake profile? I have attached the graphs (I
have not mirrored the "reverse" graphs but noshake integrates to the
same value).
I have also done a free energy of hydration for ethane to methanol
(using sander for the vacuum leg). Here I find that SHAKE is
applied in both "directions" i.e. all hydrogen atoms have SHAKE applied
but the resulting free energies differ. In the noshake case I get
~-6.4 kcal/mol which compares well with the literature but with all
SHAKE on I get something like -8 or -9. Maybe there is some wider
problem with how SHAKE is handled in softcore TI?
I can provide complete simulation results if necessary. I haven't
attached those because they are >500MB.
Many thanks,
Hannes.
-- Scanned by iCritical.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: e2m_m2e.png)
- application/octet-stream attachment: e2m_m2e.agr
