Thank you for the e mail
I checked P - TYR is in amber parameter data base .
I am interested to know about the S - TYR ,
how to proceed with this case ,
since i got a note from earlier mail that i should not use the gaff for
protein or DNA .
so if one need to derive optimized geomentary QM is must or i can use the
RESP server
i am not clear . explain me more about building the library of these kind
of amino acids
On Thu, Apr 24, 2014 at 1:45 AM, FyD <fyd.q4md-forcefieldtools.org> wrote:
> Lara,
>
> > I would like to know if P-ser , S-Tyr , P-Tyr are already deposited in
> the
> > database
>
> phosphorylated amino acid residues are available in the amber force
> field database. I wonder if they are not also in the amber
> distribution as well; to be checked...
> Sulfated-residues? I do not think so they are available.
>
> > or one have to do the partial charge derivation using QM to build the
> amber
> > force field parameters
> > is general amber gaff is enough to do that .
>
> "gaff is enough to do that" : what does that mean?
>
> I would not use gaff for proteins and nucleic acids. A force field
> designed for... proteins should be used instead; i.e. amber99SB or its
> last adaptation available in the amber distribution.
>
> regards, Francois
>
>
> > On Thu, Apr 24, 2014 at 12:38 AM, FyD <fyd.q4md-forcefieldtools.org>
> wrote:
> >
> >> Dear Lara,
> >>
> >> Do you want to generate a force field (ff) for a whole molecule or for
> >> a molecular fragment?
> >> In general one wants to generate a ff for a molecular fragment...
> >>
> >> A fragment is empirical so it is designed from a whole molecule after
> >> QM steps (geometry optimization and MEP computation) by applying
> >> specific charge constraints on group(s) of atom(s) during the charge
> >> fitting step (empirical step) and by removing the atoms involved in
> >> these constraints...
> >>
> >> You can use R.E.D. Server Development at
> >> http://q4md-forcefieldtools.org/REDServer-Development/ to generate ff
> >> for whole molecules and/or molecular fragments.
> >>
> >> In this case you start by generating a PDB file for a dipeptide for
> >> this modified amino acid (AA*); i.e. a residue with 2 peptide bonds
> >> between this AA* and two capping groups (with well defined
> >> conformation(s)): CH3CO-AA*-NHCH3 and you define charge constraints on
> >> groups of atoms to generate the wanted fragments;
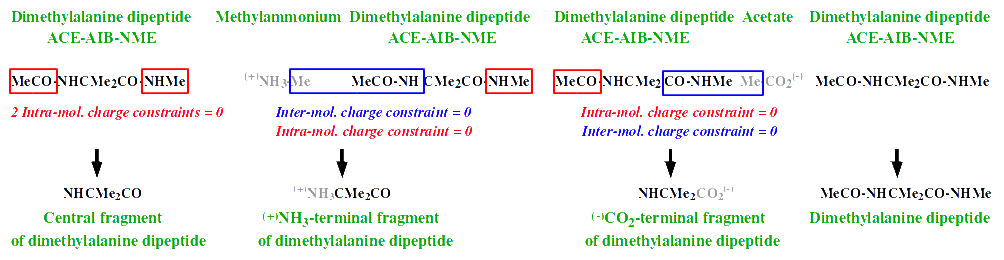
> >> see http://q4md-forcefieldtools.org/Tutorial/images2/AA-residues.gif
> >>
> >> regards, Francois
> >>
> >> ps the P2N input file format is replaced by the PDB one in
> >> REDServer-Development
> >>
> >>
> >> > I have a doubt regarding force field generation for a modified amino
> acid
> >> >
> >> > how one should take the atoms in the amino acid for running the QM .
> >> >
> >> > making this QM to build the amber force field ,
> >> >
> >> > so that the molecule is taken to the library and possibility to
> generate
> >> > the parameter for a PDB with these modified residues like it is done
> for
> >> > the default residues
> >> >
> >> > It will be helpful for me to understand if some one gives the detail
> for
> >> > the atom selections for the Qm run
>
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Wed Apr 23 2014 - 23:30:02 PDT
{kind=link}
