Thank you
I would like to know if P-ser , S-Tyr , P-Tyr are already deposited in the
database
or one have to do the partial charge derivation using QM to build the amber
force field parameters
is general amber gaff is enough to do that .
if suggestions are there please let me know
thank you
On Thu, Apr 24, 2014 at 12:38 AM, FyD <fyd.q4md-forcefieldtools.org> wrote:
> Dear Lara,
>
> Do you want to generate a force field (ff) for a whole molecule or for
> a molecular fragment?
> In general one wants to generate a ff for a molecular fragment...
>
> A fragment is empirical so it is designed from a whole molecule after
> QM steps (geometry optimization and MEP computation) by applying
> specific charge constraints on group(s) of atom(s) during the charge
> fitting step (empirical step) and by removing the atoms involved in
> these constraints...
>
> You can use R.E.D. Server Development at
> http://q4md-forcefieldtools.org/REDServer-Development/ to generate ff
> for whole molecules and/or molecular fragments.
>
> In this case you start by generating a PDB file for a dipeptide for
> this modified amino acid (AA*); i.e. a residue with 2 peptide bonds
> between this AA* and two capping groups (with well defined
> conformation(s)): CH3CO-AA*-NHCH3 and you define charge constraints on
> groups of atoms to generate the wanted fragments;
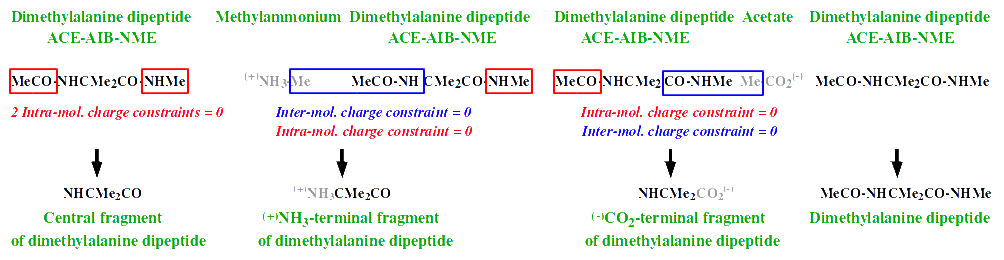
> see http://q4md-forcefieldtools.org/Tutorial/images2/AA-residues.gif
>
> regards, Francois
>
> ps the P2N input file format is replaced by the PDB one in
> REDServer-Development
>
>
> > I have a doubt regarding force field generation for a modified amino acid
> >
> > how one should take the atoms in the amino acid for running the QM .
> >
> > making this QM to build the amber force field ,
> >
> > so that the molecule is taken to the library and possibility to generate
> > the parameter for a PDB with these modified residues like it is done for
> > the default residues
> >
> > It will be helpful for me to understand if some one gives the detail for
> > the atom selections for the Qm run
>
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Wed Apr 23 2014 - 22:30:02 PDT
{kind=link}
