Date: Fri, 08 Nov 2013 17:19:40 +0800
hello,Pawel,
I am sorry to trouble you again!Thank you for your attention!
In my work, the volume rescaling was anisotropic so I think I need three
scale factor of x,y, z vector . I change the script to RevSym_mod.py
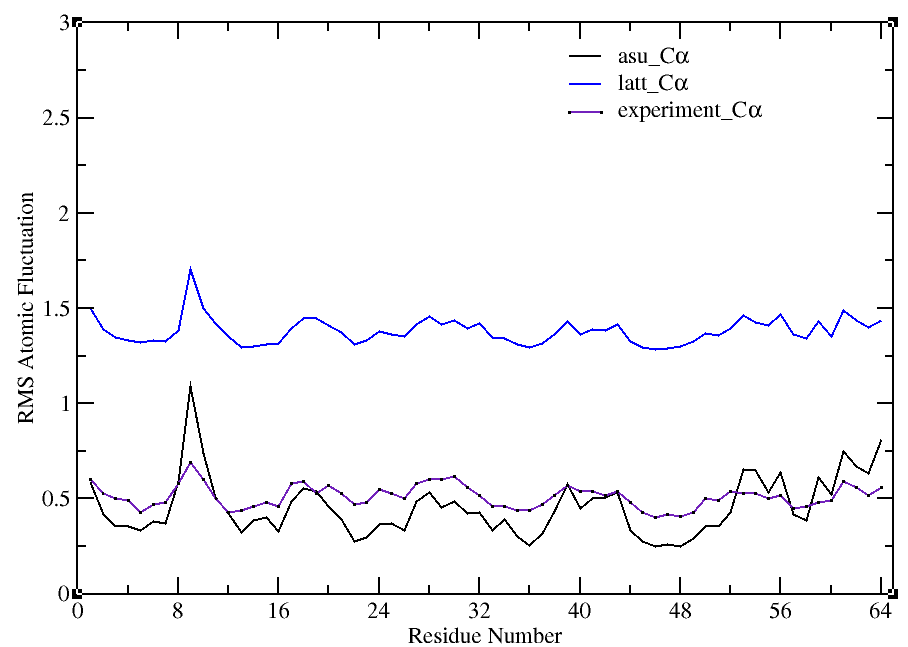
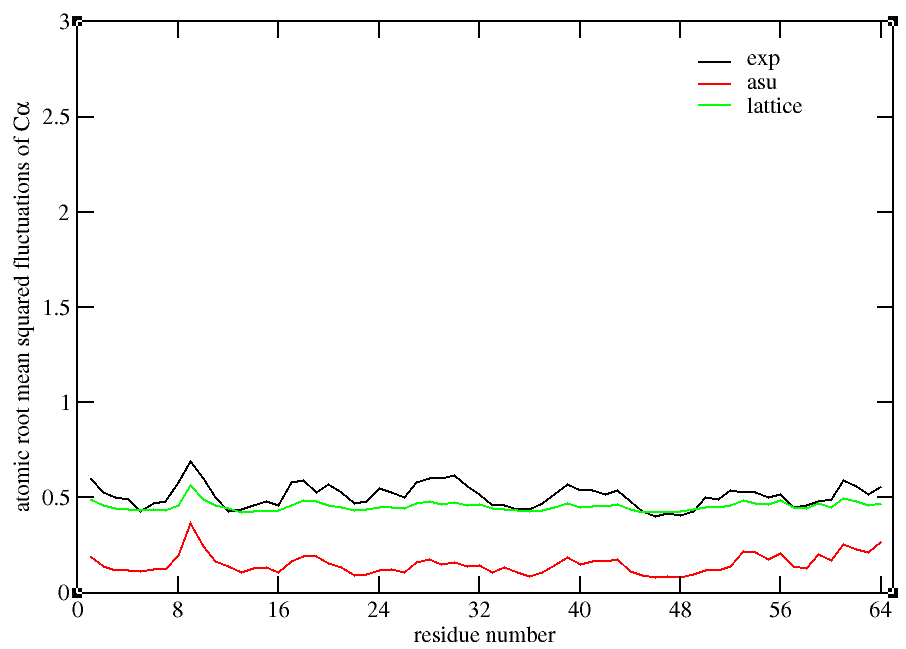
(see attachment ) .After that , the rms fluctuation of a-carbons for asu
and lattice decrease so much at once (see figure 1) but unlucky they are
so different from the result of 2010 Paper and the asu_rmsd and
lattice_rmsd become about 7.0 Å which is so high. So I want to know how
to do the least squared minimization problem which 2010 Paper used as
you said. Can you give me this script or tell me how to gain it?
Then I am confused to compute the bfactor. One method is trajin all 108
monomers trajectory and compute the bfactor (see method1) and the second
method is that trajin in each monomer trajectory and compute its bfactor
then average these 108 bfactors . We can see that the lattice_bfactor
from method2(see figure3 ) is smaller than that (see figure 2) from
method 1 but asu_bfator is bigger . So I want to know which one is right
? (BTW, before gaining the figure 2 and figure 3, we didn't do the
rescaling )
Thank you very much!
Best wishes!
Aimee Li
method1 :
#################################
# Calculate RevSymm B-factors #
#################################
f=open('ctraj_bfactor_lat','w')
f.write('parm %s\n' %topo)
for i in range(unitcells):
for j in range(asymunits):
f.write('trajin RevSym_%02d_%02d.nc\n' %(i+1,j+1))
f.write('atomicfluct out bfac_lat_calpha.dat :1-64.CA byatom bfactor\n' )
f.close()
os.system('cpptraj <ctraj_bfactor_lat')
method2:
#################################
# Calculate RotFit B-factors #
#################################
for i in range(unitcells):
for j in range(asymunits):
f=open('ctraj_bfactor_asu-bk_%02d_%02d' %(i+1,j+1),'w')
f.write('parm %s\n' %topo)
f.write('trajin ../RevSym_%02d_%02d.nc\n' %(i+1,j+1))
f.write('reference AvgCoord_asu.rst7.1\n')
f.write('rms reference mass :1-64.CA,C,N\n')
f.write('atomicfluct out bfac_asu_calpha-bk_%02d_%02d.dat :1-64.CA
byatom bfactor\n' %(i+1,j+1))
f.close()
os.system('cpptraj <ctraj_bfactor_asu-bk_%02d_%02d' %(i+1,j+1))
===============
On 10/30/2013 09:12 PM, Pawel wrote:
> Hi Aimee,
>
> To get the scaling properly you need something like this:
>
> A=40.000 #set this to the experimental value of your
> supercell's a-vector
> for frame in range(frames):
> a=celllen[frame,0] #get the a-vector of the current frame
> scale=a/A #scale factor (pressure scaling is isotropic in Amber
> so your scale factor will be the same in all dimensions
> currMoveVector=MoveVector*scale # scale your MoveVector
> coords[frame,:,:]=coords[frame,:,:]-MoveVector
>
> Try that and see what happens although if you are monitoring your volume
> and it is not deviating by more than 0.3% or so from the experimental,
> this should really not affect your B-factors all that much.
>
> Besides that I'm really not sure what the reason could be. Possibly the
> algorithm for fitting the unit cells (the least squares problem for
> finding the correct origin that I mentioned in previous e-mail) could
> have made a difference. Possibly your system is sampling an alternate
> ensemble that is producing different B-factors.
>
> Pawel
>
>
>
> On 10/29/2013 04:03 AM, Yongxiu Li wrote:
>> hello,Pawel
>> Thank you very much! I am so sorry for that I read the
>> RevSym_netcdf.py again and I think I have done the rescaling. I think
>> these lines do the all rescaling "
>> for frame in range(frames):
>> #get frame box
>> box=celllen[frame,:]
>> #convert to unit cell box
>> box=box/[ix,iy,iz]
>> #add angle information as provided by user
>> UCbox=hstack((box,SCBox[3:]))
>> #calc orthogonalization matrix
>> u,invu=CompXfrm(UCbox)
>> #matrix product
>> MoveVector=dot(invu,FracVector).astype(float32)
>> coords[frame,:,:]=coords[frame,:,:]-MoveVector
>> # reverse symmetry operate to original asym unit by
>> # applying the symmetry translation and rotation
>> coords[:,:,:]=dot( (coords[:,:,:]-t),linalg.inv(s) ) "
>> I'm not proficient in Python, so whether I understand is right? If
>> not, can you tell me how to do in python script? If I am right, I
>> still can't understand that compared with figure8 in Case's paper,why
>> the bfac_lat_calpha-arf.dat which I gained is so big.
>> btw,in the attachment is the lattice atomic root mean squared
>> fluctuations of a-carbons by amber ff99SB-tip3p.
>> Thank you very much!
>> Aimee Li
>>
>> Atomicrootmeansquared(rms)fluctuationsofa-carbons.
>>
>> On 10/16/2013 07:26 AM, Pawel wrote:
>>> Hi Aimee,
>>> On 10/15/2013 09:09 AM, Yongxiu Li wrote:
>>>> 1. if the "bfac_lat_calpha" in figure8 are calculated using a
>>>> different
>>>> approach than the one used in BasicAnalysis, how can I do it?
>>> If I understand correctly (I did not work on that paper myself), the
>>> alignment of asymmetric units was done by solving a least squares
>>> minimization problem to find (for each frame) the best location of the
>>> crystallographic origin in space so that after performing the
>>> appropriate symmetry operations and translations in reverse on each of
>>> the asymmetric units, the resulting RMSD between the asymmetric units
>>> would be minimized. Unfortunately I don't have the code for that
>>> approach. If it would be very useful to you, let me know and I'll
>>> investigate.
>>>
>>>> So as you said, I need to rescal coordinates.
>>>> However, after reading this paper I am confused that wether only
>>>> computing the lattice property (for example:rmsd_latt and
>>>> bfactor_latt)need rescal coordinates but computing the other
>>>> property(like rmsd_ASU ,bfactor_ASU and Distance deviation matrices)
>>>> needn't rescale the coordinates?
>>> If you are simulating an NPT ensemble, you should rescale the
>>> coordinates for all of these analyses. This should be done in
>>> RevSym_netcdf.py though I think the version of that script that you have
>>> does not do so. Like I said these scripts Analysis directory are all
>>> beta versions. However you should be able to do the rescaling easily by
>>> adding one or two line around line 95 of the script. If you have
>>> problems, let me know and I'll dig up a version that does this.
>>>
>>> Pawel
>>>
>>> _______________________________________________
>>> AMBER mailing list
>>> AMBER.ambermd.org
>>> http://lists.ambermd.org/mailman/listinfo/amber
>>>
>>
>>
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- text/x-python attachment: RevSym_mod.py

(image/png attachment: 3.png)
(image/png attachment: 2.png)
(image/png attachment: 1.png)
