Date: Mon, 01 Jul 2013 20:26:11 +0200
Dear prof. Case,
thank you for a prompt response !
You are right, I used iwrap=1 (in initial NVT but also in NPT production
run)
to have permanently possibility to see my system in "compact"
representation.
You are also right that the strange system "reimaging" has nothing to do
with any system physical changes, and simply iwrap function just
"decided" that it's
replication unit (simul box) will be "different" from my original system
( i.e. rather composed of several pieces of the neighboring periodic
images ) but
of course such periodic box will replicate the same infinity system as if
it
match my original system or more preciously my system in original form.
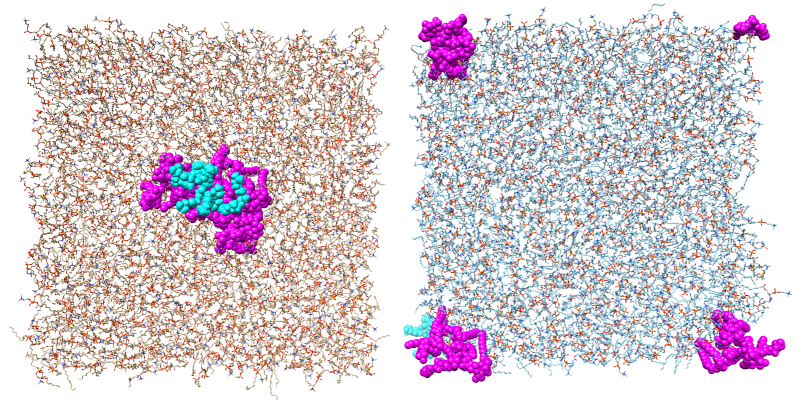
No gaps in the right part of the attached figure (the case 2 from the
previous email)
clearly demonstrates that using X,Y dimensions from the *.rst file of the
previously
equilibrated lipid bilayer was a good idea and that it works as it should.
So I was just
confused before with that strange reorganization.
BUT
#1
During the simulation (here typically some hundreds of ns) I simply can't
avoid of using
iwrap=1 (at least few times) to prevent water molecules diffuse too far
from the
original position = to prevent eventual ****** records in *.rst files.
But OK if it has no impact on simulation there is perhaps no reason to
have any problems
with iwrap=1 I guess even in case of such systems like lipid bilayers, or
am I wrong here ?
Anyway could be possible to force iwrap function (e.g. using some
additional parameter settings)
that the replication unit is the best compromise between the original
system and
the box dimensions provided ? I mean in my case to enforce it to use as
the replication unit
my system in it's original form ?
#2
At least at the end of the simulation I would like to visualize my system
in the
original form e.g. my ligand near the center of the lipid bilayer (perhaps
attaching to
the bilayer surface or penetrated into bilayer - depends on interaction
and length of the simulation), eventually below the layer 20A water shell
and above 80 A shell as I created it etc.
This (or something close to this) perhaps could be done with image action
of the ptraj/cptraj.
Perhaps no chance to achieve the original configuration so the best what i
can eventually to do
maybe center the system (map the all molecules) using the condition that
my ligand is in the
middle of the final reimaged simualtion box or so ... Am I right ?
Thanks for the additional comments !
Best wishes,
Marek
Dne Mon, 01 Jul 2013 18:12:54 +0200 David A Case
<case.biomaps.rutgers.edu> napsal/-a:
> On Mon, Jul 01, 2013, Marek Maly wrote:
>
>>
>> I am just curious if there is any recommended approach
>> how to create periodic box around solvated (externally)
>> lipid bilayer system.
>>
>> I obtained using "setbox UNIT vdw" (I used "set UNIT box { X Y Z }"
>> command in the final step).
>
> I personally don't think that setbox UNIT vdw is every very useful; the
> second
> form above is probably what you want, but of course you have to do some
> visual
> or other investigation to obtain good values for X Y Z.
>
>> Let's just notice that during the minimization phase the system was
>> maintained in a good
>> box, the problem with wrong reimaging starts from the NVT MD.
>
> Do you somehow have iwrap=1? You probably want to have no reimaging for
> these
> sorts of simulations. (Although, aside from visual appearance, it should
> work; nevertheless, I'd recommend avoiding it.)
>
> ...dac
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
> __________ Informace od ESET NOD32 Antivirus, verze databaze 8513
> (20130701) __________
>
> Tuto zpravu proveril ESET NOD32 Antivirus.
>
> http://www.eset.cz
>
>
>
-- Tato zpráva byla vytvořena převratným poštovním klientem Opery: http://www.opera.com/mail/
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: 01_02.png)
