Date: Tue, 2 Apr 2013 17:04:06 +0530
Dear amber users
I am trying to simulate a* 37* residue long *peptide u*sing *GB-OBC
*approximation.
The simulation was carried out in 3 steps-
1. Minimization
2. Heating for 1 ns from 0 to 300 K
3. Production run for 30 ns.
The inputs for the same are given below:
*minimization
&cntrl*
imin=1, maxcyc=100,
cut=300.0, igb=2, saltcon=0.2, gbsa=1,
ntpr=10, ntx=1, ntb=0,
&end
/
*Stage 1 heating of cath 0 to 300K
&cntrl*
imin=0, irest=0, ntx=1,
nstlim=500000, dt=0.002,
ntc=2, ntf=2,
ntt=3, tautp=1.0, gamma_ln=1,
tempi=0.0, temp0=300.0,
ntpr=500, ntwx=500,
ntb=0, igb=2, cut=12,
/
*Stage 2 md of cath at 30ns
&cntr**l*
imin=0, irest=1, ntx=5,
nstlim=15000000, dt=0.002,
ntc=2, ntf=2,
ntt=3, tautp=1.0, gamma_ln=1,
tempi=300.0, temp0=300.0,
ntpr=500, ntwx=500, ntwe=500, nscm=1000,
ntb=0, igb=2, saltcon=0.2, gbsa=1, cut=12,
/
~
Although the potential, total, kinetic energies, temperature when were
plotted using xmgrace, the trajectory seemed to be stable. But when the
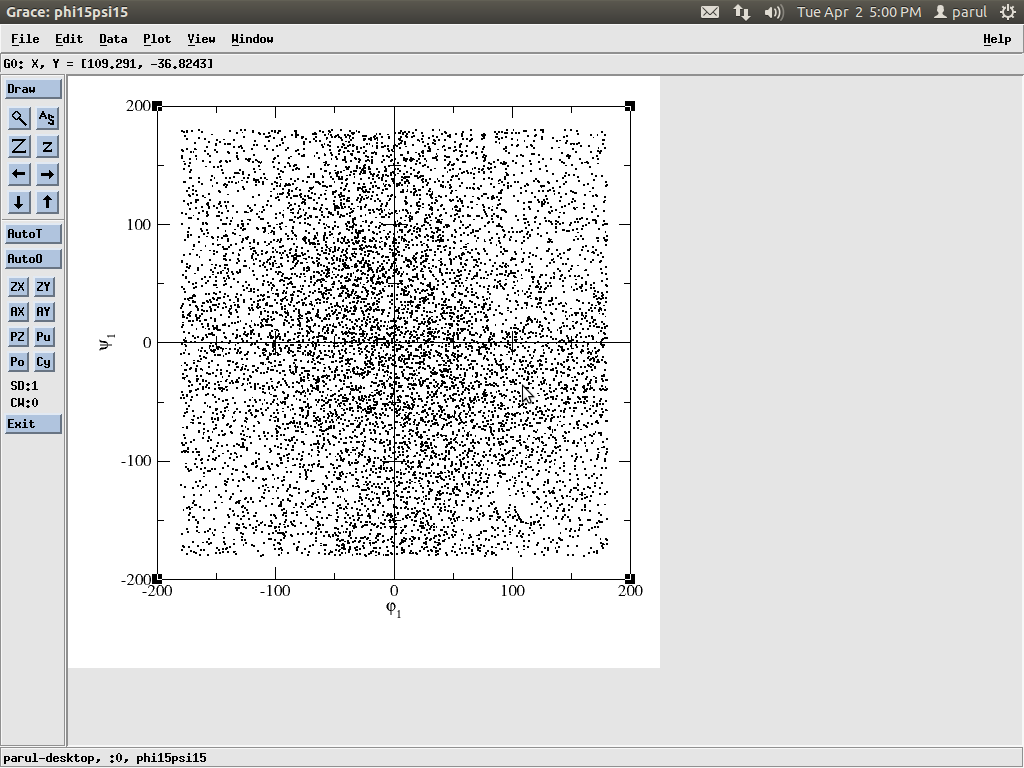
dihedrals were calculated using ptraj and were plotted using xmgrace, the
plot wasn't as expected( attached png of one dihedral angle)
When the trajectory was viewed in VMD no secondary structure is getting
attained, a *random coil* structure being shown.
Can u please help us. if some important* input paramete*r we might be
missing.
In anticipation of getting a solution from you soon.
Thanking you
Sincerely Your's
Kamakshi
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: ramachandran_plot.png)
