Date: Thu, 31 Jan 2013 14:10:55 +0800
Dear all,
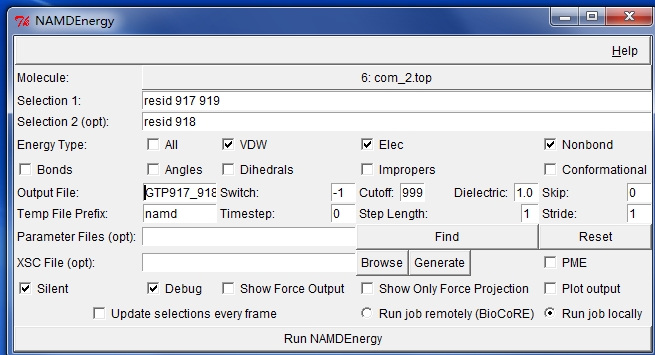
I want to calculate the pairwise interaction energy (VDW and Electrostatic) of two residues. In AMBER 11 software, MMPBSA.py script is used by using "idecomp=4" option in the input file. And In VMD 1.9.1 software, NAMDenergy plugin is used, and the force field parameter is also amber type. The detail information of parameter settings is as follow. But for the calculated results, the value of interaction energy (VDW and Electrostatic ) of NAMDEnergy is two fold of the counterpart of MMPBSA.py. I don't know whether there is difference of scaling factor between these two methods in calculating VDW and electrostatic energy. Would you like to give me some help? Thanks !
Best wishes
Yours Sincerely
Duan Baogen
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: Catch.jpg)
