Date: Tue, 29 Jan 2013 18:42:38 +0530
Dear Amber users,
I am working with the protein-ligand systems. The protein-ligand complex
structure was obtained from Protein Data Bank. Before starting the
simulation, I have carried out QM calculation ( b3lyp/6-311+g* ) of ligand
to get the optimized structure and ESP fitted point charge for the atoms.
The optimized ligand geometry was similar to the crystal structure. Then, I
have used the antechamber module of Amber to prepare the parameter file (
PREP and FRCMOD files) for the ligand, where I used the GAFF force field.
Unfortunately, the ligand geometry has been changed drastically during the
initial stages of production run ( after ~1ns of equilibration phase) of
simulation. The ligand keep its proper conformation ( i.e. close to
crystal conformation) for the initial ~1 ns phase of equilibration. Is it a
problem with force field? . If so ( i.e., if there is any problem with
ligand FF parameters), the observed conformational changes should happen in
the initial stage of the simulation rather than at the end of ~1ns
equilibration.
Can anyone help me to figure out what went wrong with my simulation.
I have attached three snapshot of the ligand (crystal structure, QM
optimized, after equilibration respectively), for your reference.
Thanks in advance
Sincerely
Aneesh
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
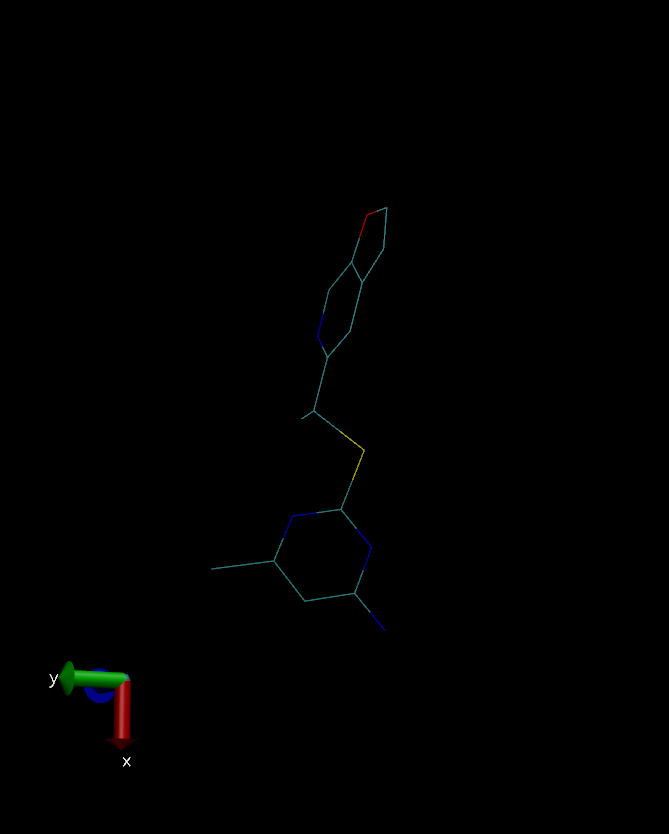
(image/png attachment: crystal.png)
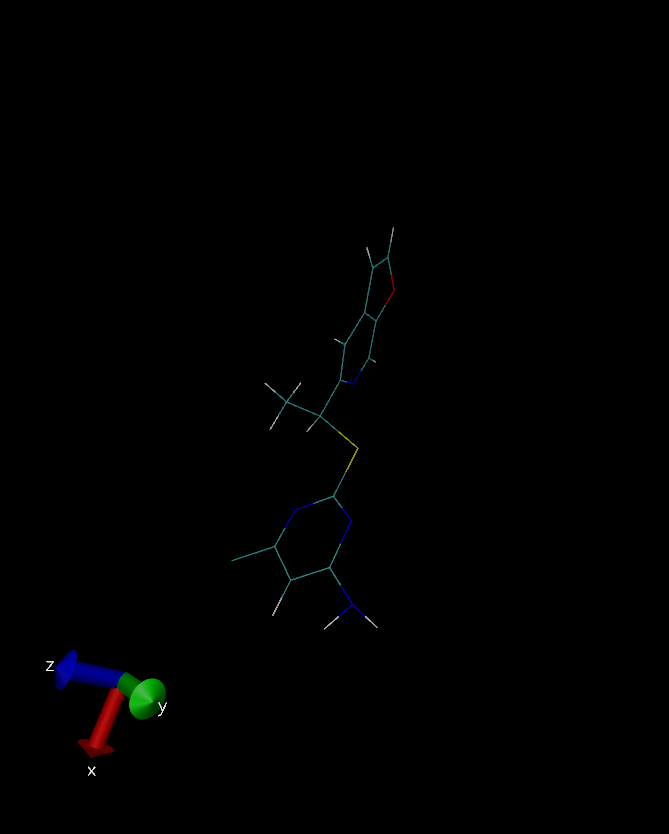
(image/png attachment: QM.png)
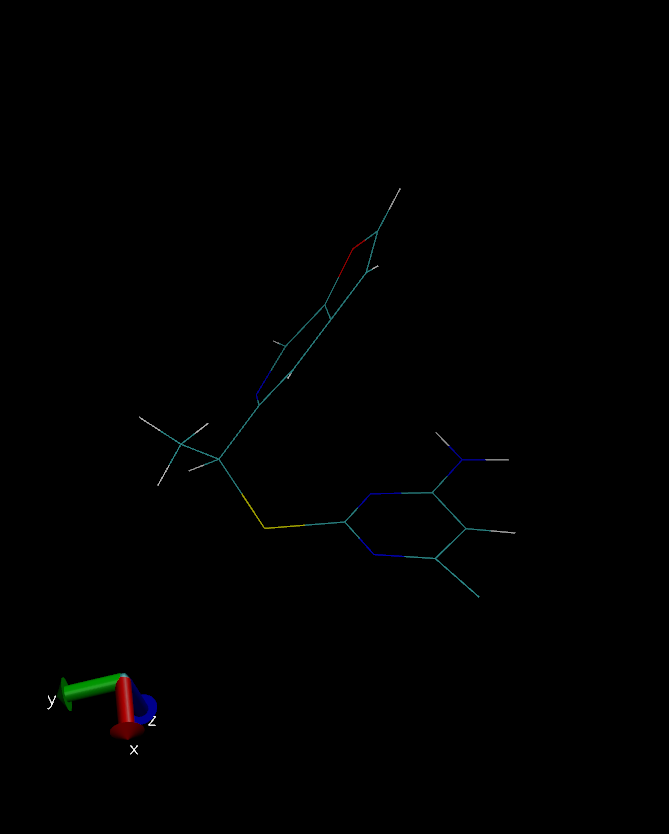
(image/png attachment: simulation.png)
