Date: Wed, 19 Dec 2012 18:20:33 +0800
Dear Lachele Foley:
Thank you for your email. It is very useful to me. ****
However, I meet some new problems. I downloaded the pdb named *2ZYK, *which
contains four gama-cyclodextrins and the water box. I ignore the other
molecular expect one cyclodextrin. Then I modify the residue name in the
pdb from *GLC* to* 4GA* so the pdb can be loaded in Leap successful and
named as *A*. ****
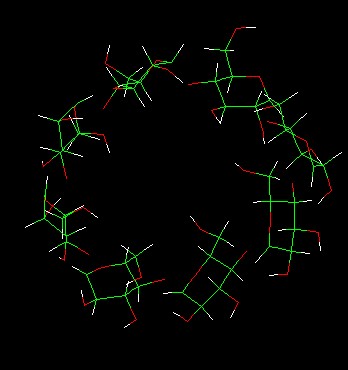
To my surprise, the molecular show a wrong structure(shown in* Fig.a*). As
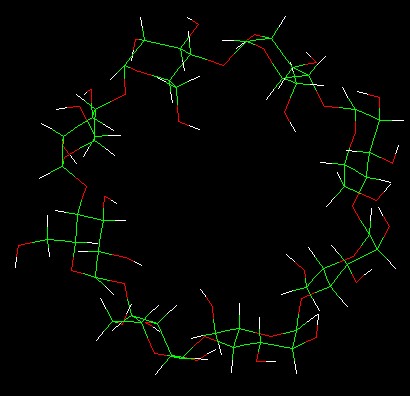
an attempt, I break all the 1,4-glycosidic bonds by typing *deletebond
A.901.21 A.902.1* and so on. The result of this showed in* Fig.b*. After
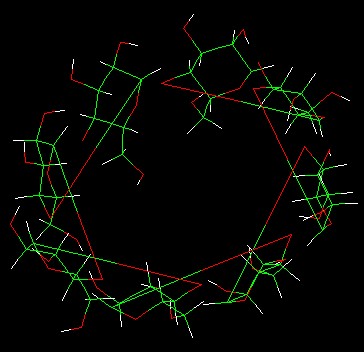
rebuild the 1,4-glycosidic bonds by typing *bond A.902.21 A.901.1*, I can
nearly get what the structure I want(*Fig.c*). However , when observing the
A.pdb gained by *savepdb A A.pdb*, I find the all the 1,4-glycosidic bonds
are breaked again, just like the *Fig.b*. ****
I get the *prmtop* and* inpcrd* file from variable *A* and use them to
create the B.pdb by *ambpdb*. This pdb performs the same mistake with the
initial pdb(*Fig.a*).****
I feel crazy. Could you tell me what’s wrong with these?****
Thanks for your help again. ****
Best wishes for you.
Fig.a
Fig .b
Fig.c
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: 3717A85D.92133C0F.4494D150.jpg)
(image/jpeg attachment: DBE5392C.92133C0F.4494D150.jpg)
(image/jpeg attachment: D5B5F662.92133C0F.4494D150.jpg)
