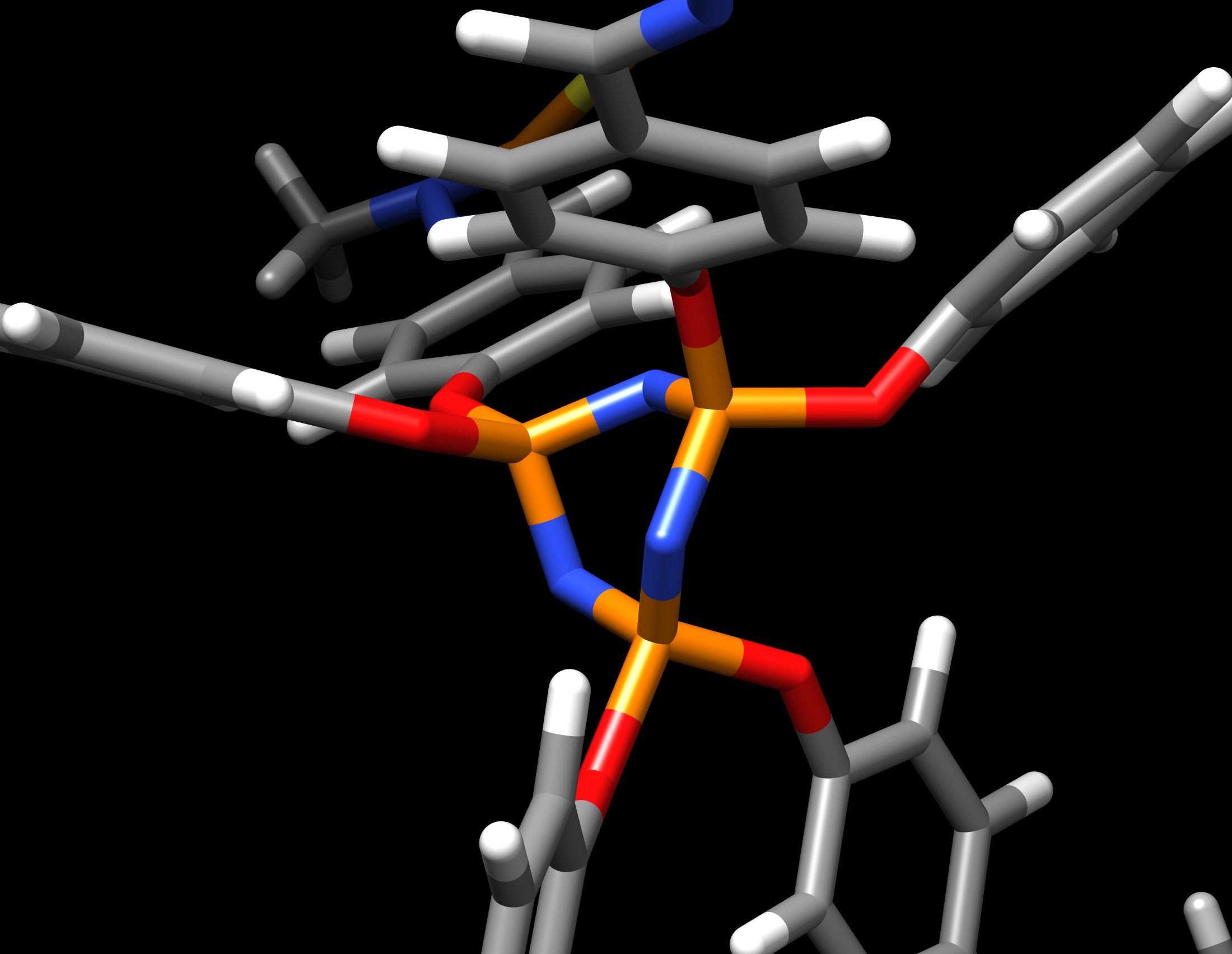
From: Giovanni Pavan <giovanni.pavan.supsi.ch>
Date: Fri, 7 Dec 2012 19:36:08 +0100
Dear all,
I am using antechamber to parametrize the cyclophosphazene ring in the
figure attached "N3P3.jpg".
Everything works fine.
Except for the planarity of the ring. In fact, this N3P3 ring should stay
planar in reality, but it does not during the simulation.
The ring bends immediately during the minimization.
By default antechamber gives "py" GAFF atom type for P. However, I have also
tried using "pd", which is supposed to be ok for aromatic P atoms.
Nothing changes.
I have also tried to increase the constant in the improper torsion,
originally:
DIHE
pb-ne-pb-ne 1 5.400 180.000 2.000 same as X
-n2-p2-X
IMPROPER
ne-pb-ne-pb 1.1 180.0 2.0 Using default
value
to:
DIHE
pb-ne-pb-ne 1 5.400 180.000 2.000 same as X
-n2-p2-X
IMPROPER
ne-pb-ne-pb 10.1 180.0 2.0 Using default
value
but, again, it didn't work - the ring bends as in figure "image.jpg".
Am I missing something?
Probably it is easy and I am making some stupid error.
What is the easiest way to have this N3P3 ring planar during the MD run?
Thanks a lot in advance,
Bye
g
Dr. Giovanni M. Pavan
Department of Innovative Technologies
University of Applied Science of Southern Switzerland
Centro Galleria 2, Manno 6928, Switzerland.
e-mail: giovanni.pavan.supsi.ch
web: http://www.dti.supsi.ch/~pavan/
skype: giovanni_pavan
phone: +41 58 666 65 60
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Fri Dec 07 2012 - 11:00:02 PST