Date: Tue, 10 Jul 2012 17:15:14 -0500
Thanks Dan,
By exploded I mean the coordinates of the atoms in the simulation are
randomly placed what looks like a cubic box with bonds criss-crossing
the box. So "exploded" in the sense that the configurations that come
out are reminiscent of a simulation in which the energy has exploded and
the molecule has gone all to heck.
The script you sent me produces the same result. Does the type of
periodic box come into play for ptraj? i.e. do I need to tell ptraj that
the box is octahedral rather than cubic? If the trajectory was smaller
(it is 17GB) I would attach it for someone else to take a crack at...
Any other thoughts on how to deal with this issue or strategies to try?
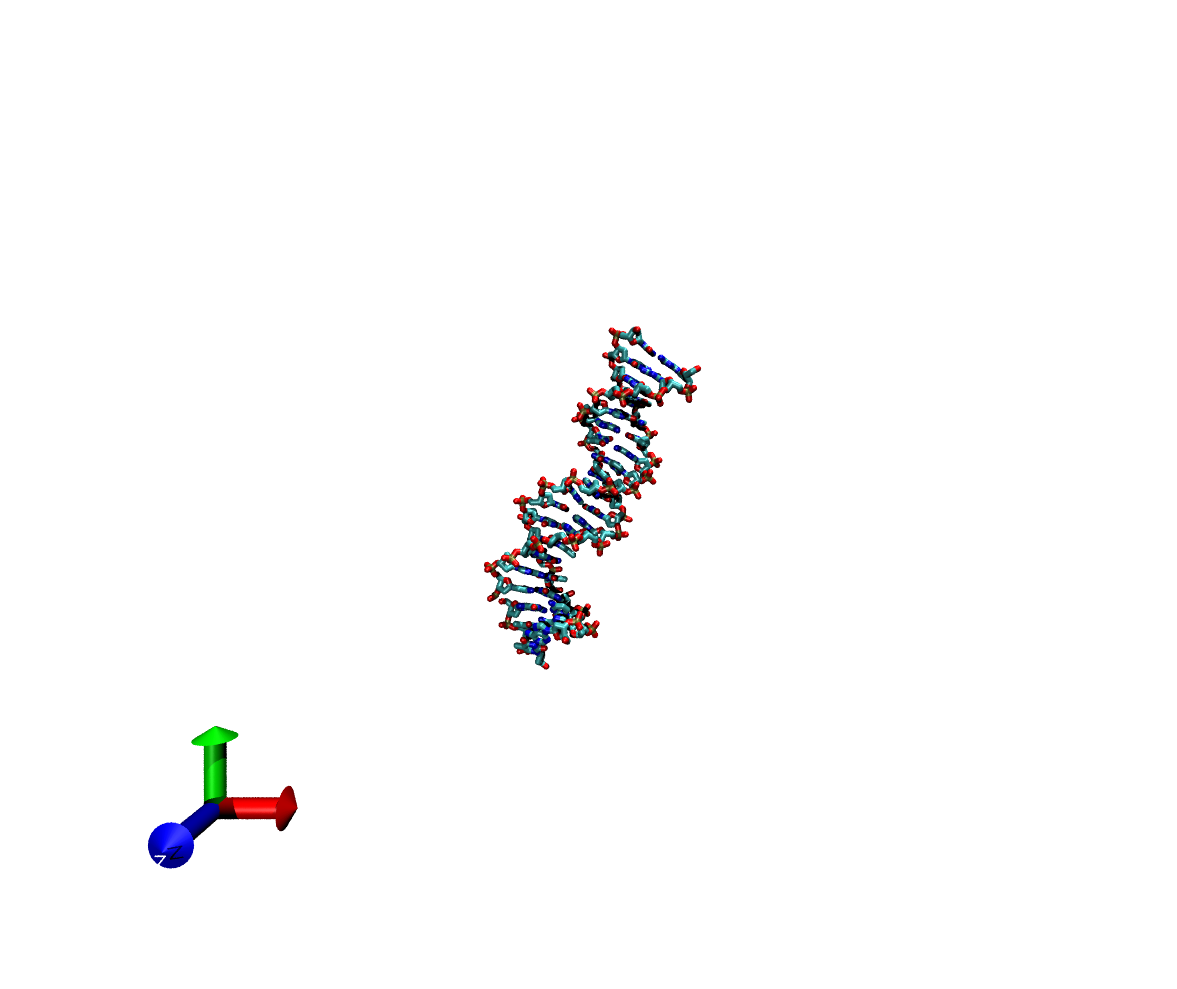
In case its helpful in explaining what I mean by "the frames look
exploded," I have attached a few vmd snapshots from early in the
simulation (looks fine) and later in the simulation (looks not so
fine)... the raw .mdcrd output looks good (like image_1.png) throughout
the simulation while the ptraj output looks distorted and "explodey."
Thanks a million,
Gordon
On 07/10/2012 09:24 AM, Daniel Roe wrote:
> Hi,
>
> Sorry for the delay in replying.
>
> So just to be clear, the script:
>
>> trajin md2.mdcrd 1000 19000 5
>> strip :WAT
>> strip :Na+
>> trajout output.pdb pdb append
> Still gives you something that looks like it exploded? By this I
> assumes you mean the two strands of DNA have separated? I think this
> is just an imaging issue. Try this:
>
> trajin md2.mdcrd 1000 19000 5
> strip :WAT,Na+
> center :1-20 mass origin
> image origin center familiar
> trajout output.pdb pdb append
>
> If you use cpptraj you can omit the 'append' keyword from the
> 'trajout' command since MODEL/ENDMDL keywords are added automatically
> for multiple frame PDB output files.
>
> -Dan
>
> On Thu, Jul 5, 2012 at 1:23 PM, Gordon S Freeman<gfreeman.wisc.edu> wrote:
>> Dear Amber Users,
>>
>> I have .mdcrd and .prmtop files that I am trying to use to generate a
>> .pdb output file that x3DNA plays nicely with. The trajectory is a 20 bp
>> DNA solvated in tleap by the solvateoct command. If I visualize the
>> .mdcrd file, everything looks just fine. I use the following ptraj
>> script to try to generate the .pdb file I need for subsequent analysis:
>>
>>
>> The resulting .pdb file looks great for the first few thousand
>> simulation frames (i.e. frames 1000 to almost 5000 from the simulation
>> convert just fine) while the latter 75% of the frames produce .pdb
>> structures that look like a cubic periodic box that has exploded. The
>> energies and .mdcrd file look fine so an explosion is clearly not the
>> explanation. Am I missing something in my ptraj input file above to
>> properly deal with the non-cubic periodic box the simulation was run
>> with? Does anyone have a script they have successfully used to do this
>> conversion? I could easily generate the pdb using VMD and the
>> correct-looking mdcrd file and then write a script to add the MODEL and
>> ENDMDL flags 3DNA looks for but I'd rather get my ptraj script working
>> properly.
>>
>> FYI, using the same script above without the lines "unwrap :1-40\ center
>> :1-40 mass origin\ image origin center familiar\" also produces the same
>> strange output.
>>
>> Thanks in advance for this and all the other help this listserve and the
>> developers have provided over the years.
>>
>> -Gordon
>>
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image_1.png)

(image/png attachment: image_2.png)
