Date: Wed, 28 Sep 2011 19:10:52 +0100
My final and positive email on this subject.
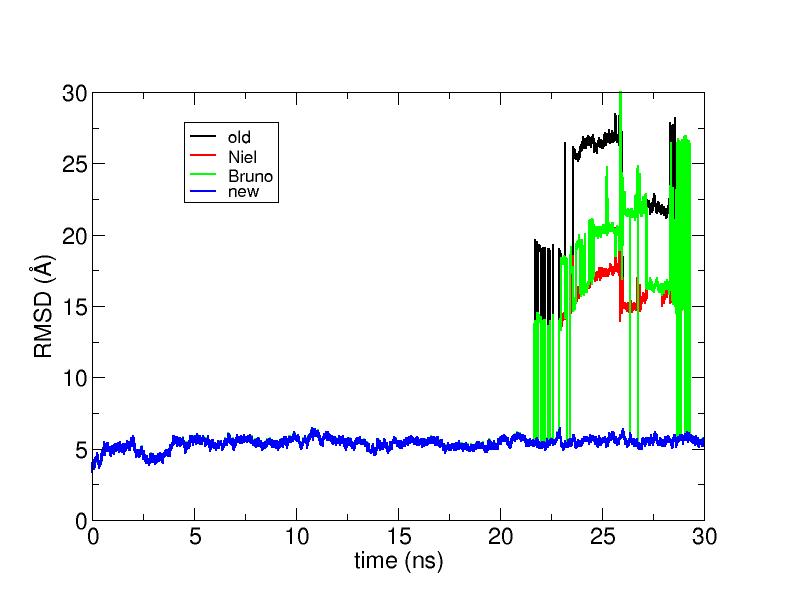
I found out that the problem might be either in the NAMD dcd
trajectory file format
or in the way trajin reads it.
If I convert the DCD file in Amber file (mdcrd) format first and then
I execute the commands suggested by Niel and Carlos I get
a "canonical" RMSD time series (solid blue line in the attached graph):
(1)
trajin tetra_dyn.dcd
trajout tetra_dyn.mdcrd
(2)
trajin tetra_dyn.mdcrd
center :1-11 mass origin
image origin center familiar
center :1-22 mass origin
image origin center familiar
center :1-33 mass origin
image origin center familiar
center :1-44 mass origin
image origin center familiar
reference tetra.pdb
rms reference out rmsd.dat :1-44.C,CA,N time 2
trajout tetra_dyn.imaged.mdcrd
And also, in displaying the imaged trajectory tetra_dyn.imaged.mdcrd,
this time I get the
whole solute (tetramer) in the middle of my truncated octahedron box for the
entire simulation.
Many thanks again for your helps and useful suggestions.
Best,
Il 28 settembre 2011 12:26, Massimiliano Porrini <M.Porrini.ed.ac.uk>
ha scritto:
> Dear Carlos, Tom and Bruno,
>
> From what I understood by "playing" with my trajectory and your very
> useful comments I should probably increase the size of my cell
> (and this time I will hack the topology file so that to have only
> one molecule as solute).
>
> If I display the trajout imaged trajectory, at some point, I still see
> one monomer
> separated from the three other monomers, hence I assume that
> Tom and Bruno are right: my cell is too small.
>
> Using a cut off equal to 10 Angs I thought that a closest distance of 11 angs
> would have been appropriate for the physical behaviour of my system:
>
> solvateoct tetramer TIP3PBOX 11.0
>
> But apparently this box is too small.
>
> Another thing I should probably point out is that I generated the trajectories
> with NAMD (using Amber coordinates and topology files), anyway I carefully
> checked my settings through the following page:
>
> http://ambermd.org/namd/namd_amber.html
>
> And what I derived myself are the x, y and z values for NAMD
> cellOrigin keyword through
> these commands in VMD:
>
> % set sel [atomselect top "all"]
> % measure center $sel
>
> i. e. I assumed that the origin of the cell is equivalent to the geometric
> center of my system, am I wrong in this? Could this be the problem of
> re-imaging?
>
> Thanks a lot for your hints.
>
> All the best,
>
>
>
> Il 27 settembre 2011 22:39, Bruno Rodrigues <bbrodrigues.gmail.com> ha scritto:
>> What happens when you load the wrapped trajectory, like in Carlos' script?
>> What does really happen with the solute. It can then give a good indication
>> about what is wrong. Does one or two strands go to the next cell? Or does
>> the system has any kind of sharp denaturation (probably not, but we never
>> know...)?
>>
>> I've learned some stuff from my work with RMSD, and it sounds that a part
>> of your molecule really went to the next cell. And I tell you more, the
>> distance between 2 cells is around 20-25 angstroms, which means that you
>> have a 12 angstroms solvent layer around your solute. I would say that it's
>> too few for a 44 residues molecule. I use 12 angstroms for a 20 residues.
>> Maybe someone more experienced can tell something more.
>>
>> On Tue, Sep 27, 2011 at 6:14 PM, Carlos Simmerling <
>> carlos.simmerling.gmail.com> wrote:
>>
>>> to follow up on Tom's last sentence ("make it so all the molecules are
>>> together in the prmtop"), here is a bit of info on how to do it:
>>>
>>> Modify the pointers in your prmtop so that all the solute molecules are in
>>> 1
>>> "molecule"
>>>
>>> - here is a sample prmtop for a dimer- the goal is to put both monomers
>>> into a single "molecule"
>>>
>>>
>>> %FLAG SOLVENT_POINTERS
>>> %FORMAT(3I8)
>>> 830 10924 3
>>>
>>>
>>> -
>>> - this data is really in this format
>>>
>>>
>>> FORMAT(12I6) IPTRES, NSPM, NSPSOL
>>> IPTRES : final residue that is considered part of the solute,
>>> reset in sander and gibbs
>>> NSPM : total number of molecules
>>> NSPSOL : the first solvent "molecule"
>>>
>>>
>>> -
>>> - what you need to do is to
>>> - leave IPTRES alone (your solute is not changing)
>>> - change NSPM to match your "new" # molecules (if you make a dimer
>>> into a single molecule, subtract 1. if you make 3 molecules
>>> into 1, subtract
>>> 2).
>>> - change NSPSOL the same way- subtract 1 or 2 or more, depending on
>>> what you subtracted from NSPM
>>> - here is the "new" SOLVENT_POINTERS section that turns the 2 monomers
>>> in a single molecule:
>>>
>>>
>>> %FLAG SOLVENT_POINTERS
>>> %FORMAT(3I8)
>>> 830 10923 2
>>>
>>>
>>> - now for the next section to change- atoms per molecule.
>>>
>>>
>>> %FLAG ATOMS_PER_MOLECULE
>>> %FORMAT(10I8)
>>> 6015 6015 3 3 3 3 3 3 3
>>> 3
>>> 3 3 3 3 3 3 3 3 3
>>> 3
>>>
>>>
>>> -
>>> - what you want to do is combine the first two "molecules" (the
>>> monomers) into a single molecule.
>>> - change the "atoms per molecule" to combine the atoms from the two
>>> molecules
>>> - in this case, 6015+6015=12030
>>> - move the numbers up- in this case, change the second 6015 to a 3,
>>> and delete the last 3 at the end of this section.
>>> - here is the modified section (not showing the removal of the final 3
>>> from the end of this section- bu make sure you delete it).
>>>
>>>
>>> %FLAG ATOMS_PER_MOLECULE
>>> %FORMAT(10I8)
>>> 12030 3 3 3 3 3 3 3 3
>>> 3
>>> 3 3 3 3 3 3 3 3 3
>>> 3
>>>
>>>
>>> - *SAVE THIS TO A NEW FILE NAME, MAKING IT CLEAR BY THE NAME THAT YOU
>>> MODIFIED THE MOLECULE POINTERS*
>>>
>>>
>>> On Tue, Sep 27, 2011 at 4:55 PM, Thomas Cheatham III <tec3.utah.edu>
>>> wrote:
>>>
>>> >
>>> > > I try to resend my email that probably was not read/received.
>>> > > Any hint to fix my RMSD values would be really appreciated.
>>> >
>>> > There is probably not an easy way to fix. Look through the archive as
>>> > suggested about imaging issues and try things like center only the first
>>> > residue of each chain, etc. and imaging. Imaging based on the first atom
>>> > rather than center of mas, etc.
>>> >
>>> > This was what prompted the question on the reflector back to you about
>>> box
>>> > size / amount of solvent. Likely there is too little, so there is not an
>>> > easy way to, by generic procedure, image the molecules back since the
>>> > periodic image may be closer than the unit cell, i.e. once it is wrapped,
>>> > it becomes closer to its image.
>>> >
>>> > I have been meaning to write a smarter imaging routine that would attempt
>>> > to minimize distances, however I have not done this.
>>> >
>>> > There are ways to make it so all the molecules are together in the prmtop
>>> > which will prevent this.
>>> >
>>> >
>>> > --tec3
>>> >
>>> > _______________________________________________
>>> > AMBER mailing list
>>> > AMBER.ambermd.org
>>> > http://lists.ambermd.org/mailman/listinfo/amber
>>> >
>>> _______________________________________________
>>> AMBER mailing list
>>> AMBER.ambermd.org
>>> http://lists.ambermd.org/mailman/listinfo/amber
>>>
>>
>>
>>
>> --
>> --
>> Bruno Barbosa Rodrigues
>> PhD Student - Physics Department
>> Universidade Federal de Minas Gerais - UFMG
>> Belo Horizonte - Brazil
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>>
>
>
>
> --
> Dr Massimiliano Porrini
> Institute for Condensed Matter and Complex Systems
> School of Physics & Astronomy
> The University of Edinburgh
> James Clerk Maxwell Building
> The King's Buildings
> Mayfield Road
> Edinburgh EH9 3JZ
>
> Tel +44-(0)131-650-5229
>
> E-mails : M.Porrini.ed.ac.uk
> mozz76.gmail.com
> maxp.iesl.forth.gr
>
-- Dr Massimiliano Porrini Institute for Condensed Matter and Complex Systems School of Physics & Astronomy The University of Edinburgh James Clerk Maxwell Building The King's Buildings Mayfield Road Edinburgh EH9 3JZ Tel +44-(0)131-650-5229 E-mails : M.Porrini.ed.ac.uk mozz76.gmail.com maxp.iesl.forth.gr
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: rmsd.jpg)
