Date: Mon, 26 Sep 2011 15:46:52 +0100
Dear Niel and dear all,
Thanks very much for the hint, but, unless I am missing something here,
unfortunately it did not work in my case.
As I think my problem is exactly the same as the one presented in the post
suggested by Niel, I updated my script commands:
trajin tetra_dyn.dcd
center :1-11 mass origin
image origin center familiar
center :1-22 mass origin
image origin center familiar
center :1-33 mass origin
image origin center familiar
center :1-44 mass origin
image origin center familiar
reference tetra.pdb
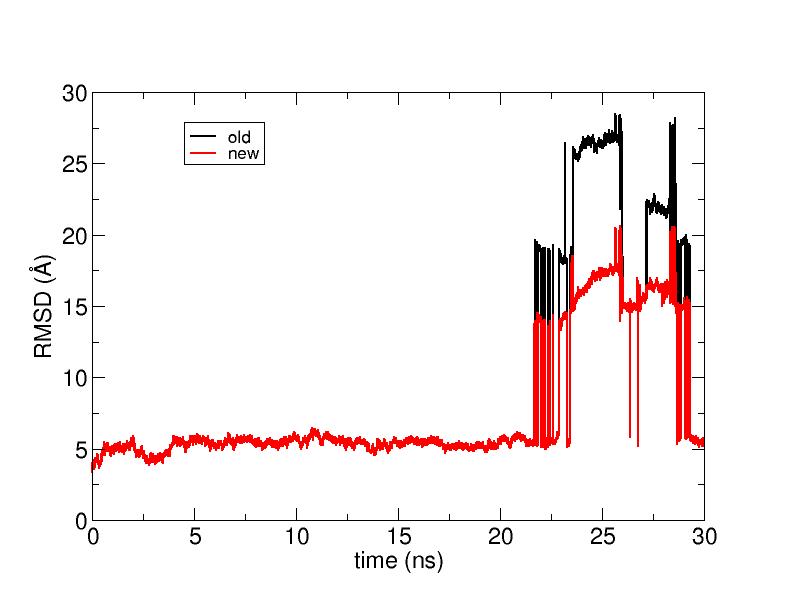
rms reference mass out rmsd.dat .C,CA,N time 2
And as you can see from the attached graph the RMSD has slightly
improved, but still I get those jumps at the end.
Any idea of what's happening here would be really appreciated.
All the best,
Il 25 settembre 2011 16:47, Niel Henriksen <niel.henriksen.utah.edu> ha scritto:
> The Amber mailing list archive is a great resource to search:
> http://archive.ambermd.org/
>
> It so happens that this issue has been discussed numerous times.
> It is an imaging issue ... this post explains what to do nicely:
> http://archive.ambermd.org/201103/0607.html
>
> If you search the archive there are lots of examples and a few
> different approaches to dealing with it.
>
> Good luck,
> --Niel
> ________________________________________
> From: Massimiliano Porrini [M.Porrini.ed.ac.uk]
> Sent: Sunday, September 25, 2011 9:22 AM
> To: AMBER Mailing List
> Subject: [AMBER] fixing rmsd values
>
> Dear all,
>
> I have worked out the rmsd time series for a 30 ns trajectory
> of my system, which is a tetramer of a 11-residues peptide
> in explicit water, using the following ptraj commands:
>
> trajin tetra_dyn.dcd
> center :1-44 mass origin
> image :1-44 origin center
> reference tetra.pdb
> rms reference mass out rmsd.dat .C,CA,N time 2
>
> As you can see from the attached graph the rmsd time series bears
> some "jumps".
> These are due to the fact that the center of mass of one or more monomers
> exceeds the boundaries of my cell (truncated octahedron) during the
> simulation and
> generate the high values of the RMSD.
>
> Does anybody know how I might fix the RMSD values via any ptraj commands?
> Any suggestion would be really appreciated.
>
> Best,
>
> --
> Dr Massimiliano Porrini
> Institute for Condensed Matter and Complex Systems
> School of Physics & Astronomy
> The University of Edinburgh
> James Clerk Maxwell Building
> The King's Buildings
> Mayfield Road
> Edinburgh EH9 3JZ
>
> Tel +44-(0)131-650-5229
>
> E-mails : M.Porrini.ed.ac.uk
> mozz76.gmail.com
> maxp.iesl.forth.gr
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- Dr Massimiliano Porrini Institute for Condensed Matter and Complex Systems School of Physics & Astronomy The University of Edinburgh James Clerk Maxwell Building The King's Buildings Mayfield Road Edinburgh EH9 3JZ Tel +44-(0)131-650-5229 E-mails : M.Porrini.ed.ac.uk mozz76.gmail.com maxp.iesl.forth.gr
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: rmsd.jpg)
