Date: Mon, 19 Sep 2011 13:34:54 +0800
Dear Amber users,
I want to unfold my protein by heating it up to 400K, then following by MD
run to expect it re-fold again. I got some results (png attached), but Im
not sure that it was unfolded with that temperature.
I viewed the results using VMD, but I got confused with the differences of
using *rst* and *crd* files.
The *rst* showed me the "full" structure while the *crd *showed me the
"scattered" structure, especially in the MD frames.
Could anyone please suggest which files I should use if I want to make sure
that my structure got unfoled and folded again by using visualization? My MD
for re-folding is still running.
Btw, I found a related issue in AMBER forum, and Jason said that "*The
restart is actually a full time step ahead with the coordinates (and a **half
time step ahead for velocities)", may I have it more clearer?
Thank you for any help.
Chinsu
** *
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
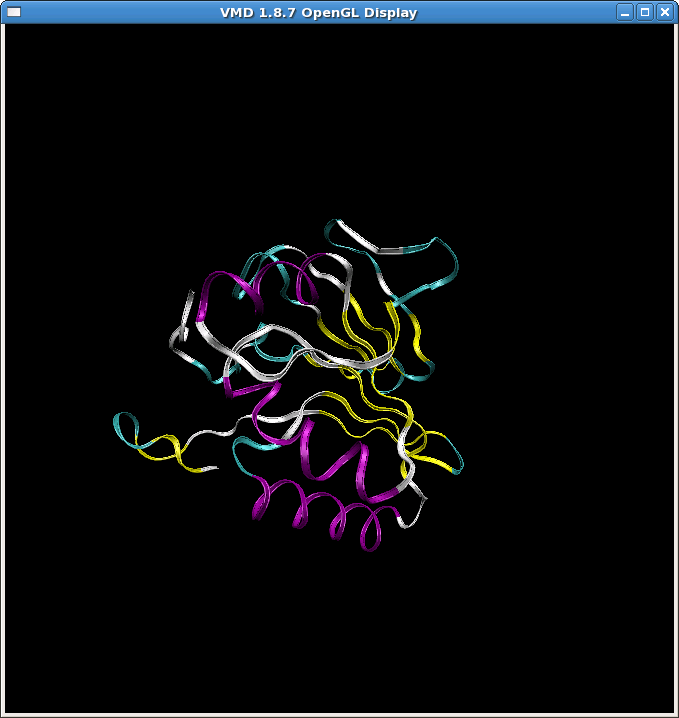
(image/png attachment: min2.png)
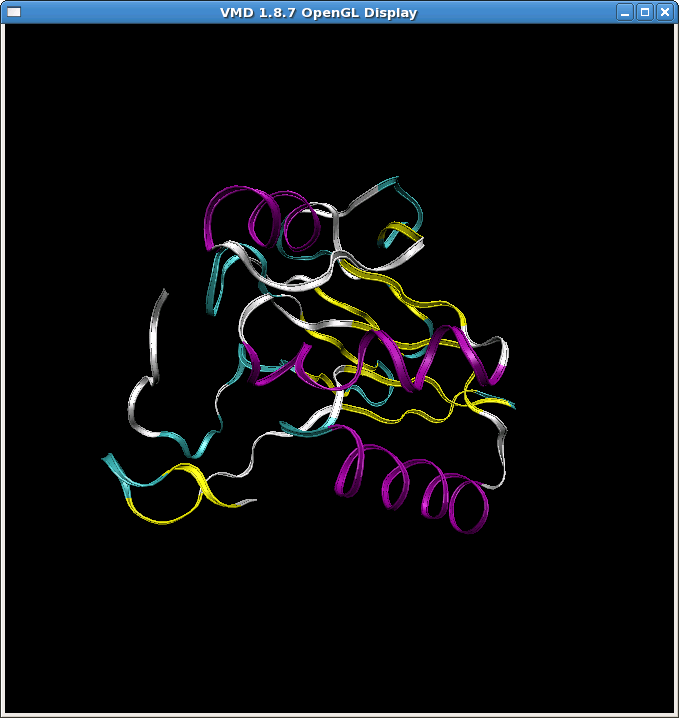
(image/png attachment: heat7.png)

(image/png attachment: heat7_crd.png)
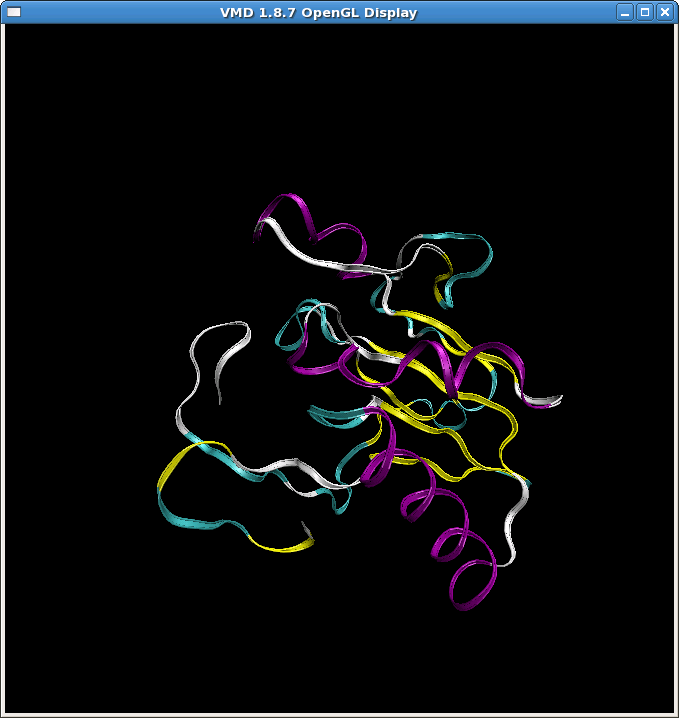
(image/png attachment: md1.png)

(image/png attachment: md1_crd.png)

(image/png attachment: md1_crd_zoom.png)
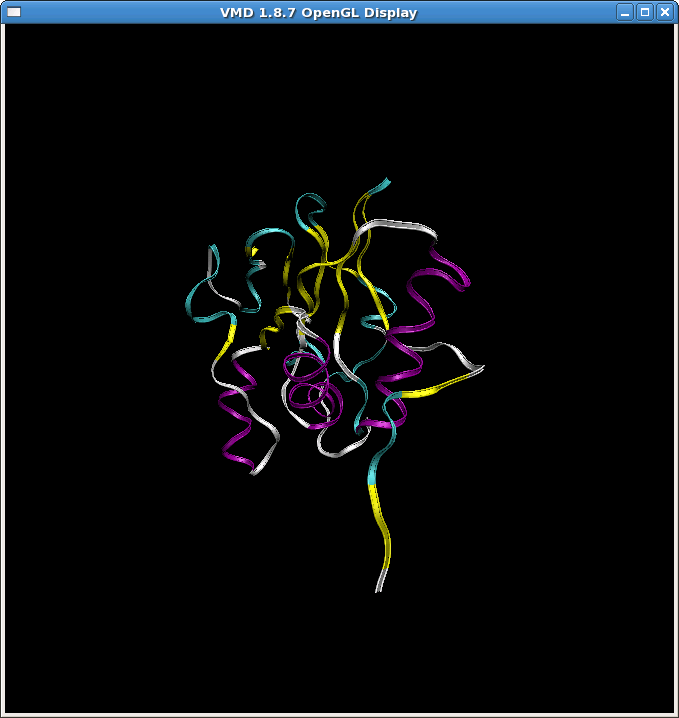
(image/png attachment: md11.png)

(image/png attachment: md11_crd_zoom.png)
