Date: Fri, 26 Aug 2011 18:16:11 +0800
Hi AMBER users,
I have some doubts, could you please help me to explain them?
I run a 20ns MD simulation for my protein (158 aa) to detect the protein
folding process. The protein was modeled using a template (homology
modeling) before running MD.
Firstly, I ran a short minimization for the protein, then a heating process
was conducted in 20ps, and followed by a 10-stage MD run (2ns for each).
I got the result, and extracted the lowest energy structure at 6.934 ns
(frame 457 of the md4.mdcrd). *Does this mean that my protein obtained the
folded state at this point of time or it just was trapped in some local
minima? *because at time point 6.934ns is it very "early" compared to the
whole process of 20ns?
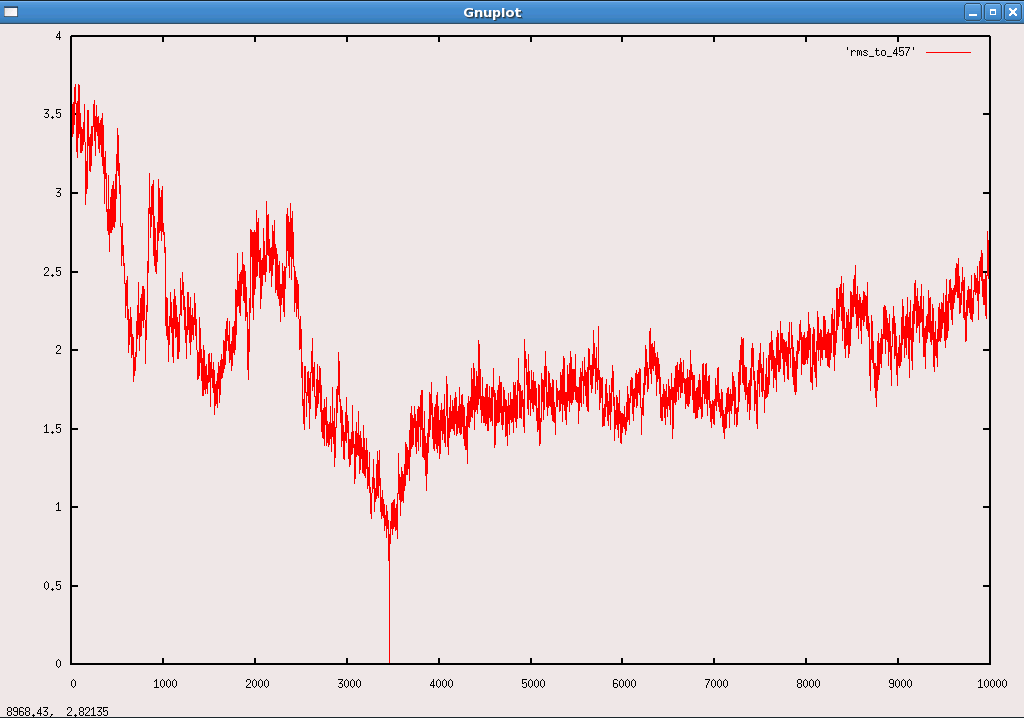
Then I plotted the backbone_RMS of the 10 stage results (compared to the
lowest energy structure). The plot was attached here too. It turned out that
there were some other time points showing stable RMS fluctuations too. As
referred to the tutorial, *does the result show that the protein kept
folding and unfolding during the time?*
*
*
And one more question is how come the plot only showed up with 10,000 ps in
the x-axis.
*trajin ../ndm_md1.mdcrd
trajin ../ndm_md2.mdcrd
trajin ../ndm_md3.mdcrd
trajin ../ndm_md4.mdcrd
trajin ../ndm_md5.mdcrd
trajin ../ndm_md6.mdcrd
trajin ../ndm_md7.mdcrd
trajin ../ndm_md8.mdcrd
trajin ../ndm_md9.mdcrd
trajin ../ndm_md10.mdcrd
reference E_lowest_457.pdb
rms reference out rms_to_457 .N,CA,C time 1.0
*Thank you very much for any help.
Regards,
Chinsu
**
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: rms_to_457.png)
