From: Hoshin Kim <85hskim.gmail.com>
Date: Thu, 2 Dec 2010 13:34:44 +0900
Dear Amber user,
I'm trying to do simulation regarding Enzyme in various solvents using
AMBER11
About one month ago, I asked for peak increasing problem at specific time.
After received replies, I applied the bugfixes with recent version, changing
some parameters in input scripts(add an ioutfm=1, delete scee) but, error is
still occurred. (Problem which I am struggling with is attached at the very
end)
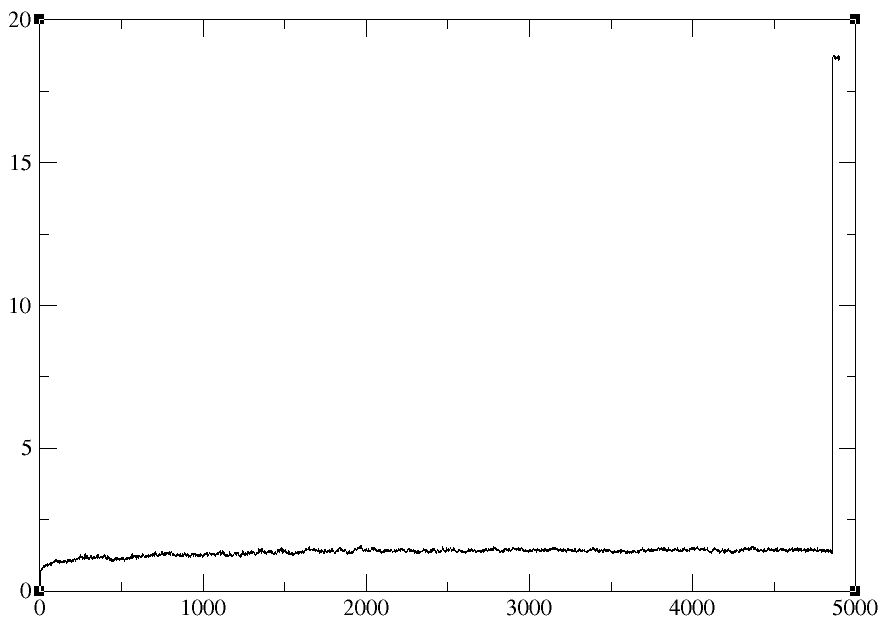
I found something strange. I have 3 split trajectory files which have 2ns
production run each(total 5ns simulation). When I merge these files as
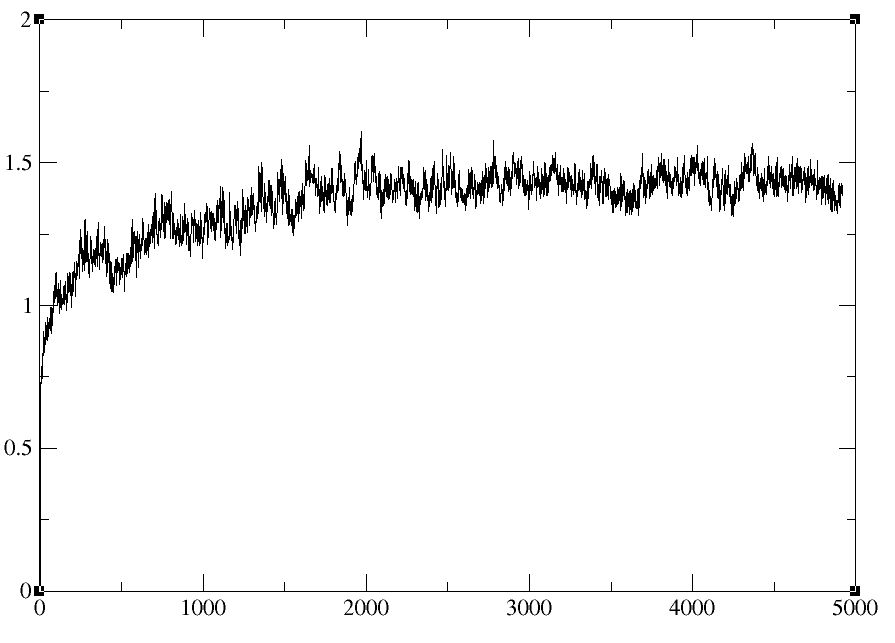
single trajectory using PTRAJ 11 then analyze RMSd, the Peak is still soared
at specific time (Case1.jpg). Protein structure is also collapsed at that
time (I think they lost their periodic boundary information). But when the
trajectory files are used directly to calculate the RMSd plotting , there
are no problem (Case2.jpg). And when I look at the trajectory which has time
point observed serious problem, structure are not lost at that time
anymore.
I could analyze RMSd and RMSf, something like that, but I'm afraid because
of this error. Could anyone please suggest me what is going on?
Thanks in advance
Hoshin Kim
<Scripts for case1 - problem >
First, I merged files into one (sometimes, I run this script with center
command)
trajin md.x
trajin md2.x
trajin md3.x
trajout md_5ns.x trajectory
then, analyze RMSd
trajin md_5ns.x
rms .
<Scripts for Case2 - no problem)
Trajectory files are directly used to analyze rmsd
trajin md.x
trajin md2.x
trajin md3.x
rms mass out 123.xmgr :1-317
----------------------------------------------------------------------------
-------
Hoshin Kim
Now)
Visiting Scholar
Department of Chemical and Biomolecular Engineering
North Carolina State University
Raleigh, NC 27695
Phone: 919-986-9258
Also)
Graduate student (M.D. candidate)
Ionic Liquids Team(ILs) in Bio Nano Process Lab
Department of Biological Engineering,
2S 113, Engineering building Inha University,
253 Yonghyeon Dong, Nam Ku
Incheon, KOREA
TEL) +82-32-860-8655
----------------------------------------------------------------------------
-------
_____________________________________________
From: Hoshin Kim [mailto:85hskim.gmail.com]
Sent: Wednesday, November 10, 2010 5:40 PM
To: amber.ambermd.org
Subject: Serious problem at specific time point
Dear Amber user, << File: K-20101110-620767.jpg >> << File:
K-20101110-617463.jpg >>
I'm trying to do simulation about Enzyme in various solvents, using AMBER11.
But I have a serious problem on my trajectory.
When I calculated RMSd of catalytic domain of enzyme, slope was suddenly
soared at 4.8ns (I attached a plotting data). So I ran the VMD to visualize
what happened at this point, as I attached, protein structure was suddenly
collapsed at that point !
This is my second try. This problem was occurred at almost same point when I
tried to do first simulation using same enzyme, same protocol. So I think
this error is not the cause of hardware, or something like that
I'm doing one more simulation using different solvent, It has also same
problem. (Peak is suddenly increased at specific time-steps) only difference
is time point.
Could anyone figure out this serious problem?
Thanks in advance,
Hoshin Kim,
P.S.
1.Attached files are only about the problem which I explained first.
2.First case, solvent is 0.3M NaCl solution, Second case, [Bmim][TfO]
3.I am using enzyme named 1TCA (CalB), and protocol for production MD is as
follows,
-------------------------------------------------------------
imin=0,
ntxo=1, ntrx=1,
cut=9.0, tempi=300.0, ntwprt=0,
ntpr=500, ntwx=500, ntwe=500, ntwr=500,
nstlim=10000000, temp0=300.0,
dt=0.002, nscm=100,nsnb=10,dielc=1.0,
ntc=2, ntf=2, tol=0.00001,
ntx=5, irest=1, heat=0.0,
ntb=2, ntp=1, pres0=1.0, taup=0.5,
scee=1.2, dt=0.002,
ntt=1, vlimit=20.0,comp=44.6,
ig=71277,
&end
-----------------------------------------------------
If further protocols are need to figure out this problem (Protocols for
minimization, equilibration ...), please tell me !
----------------------------------------------------------------------------
-------
Hoshin Kim
Now)
Visiting Scholar
Department of Chemical and Biomolecular Engineering
North Carolina State University
Raleigh, NC 27695
Phone: 919-986-9258
Also)
Graduate student (M.D. candidate)
Ionic Liquids Team(ILs) in Bio Nano Process Lab
Department of Biological Engineering,
2S 113, Engineering building Inha University,
253 Yonghyeon Dong, Nam Ku
Incheon, KOREA
TEL) +82-32-860-8655
----------------------------------------------------------------------------
-------
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Thu Dec 02 2010 - 11:00:04 PST