Date: Wed, 26 May 2010 17:11:45 +0430
Hello all,
I have problem with adjusting the number of minimization steps for my MD
simulation.
I ran 3 MD simulations in explicit solvent with a protein containing 181
a.a. for 2 nanosec and I got
3 different sets of data in which the RMSd, RMSf, radius of gyration and
other data were a bit different in each one!!
Minimization steps were adjusted as below:
run #1 : 2500 steps (1000steps steepest decent+1500 steps conjugate
gradient)
run #2: 5000 steps (2000 steps steepest decent+3000 steps conjugate
gradient)
run #3 :10000 steps (2000 steps steepest decent+8000 steps conjugate
gradient)
Is it normal to obtain different sets of data upon doing different
minimization steps?
How many minimization steps should be sufficient? How is it possible to find
ot its suficiency?
Data related to radius of gyration, rmsd and rmsf are attached as pictures.
Thanks in advance,
M. Reza
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
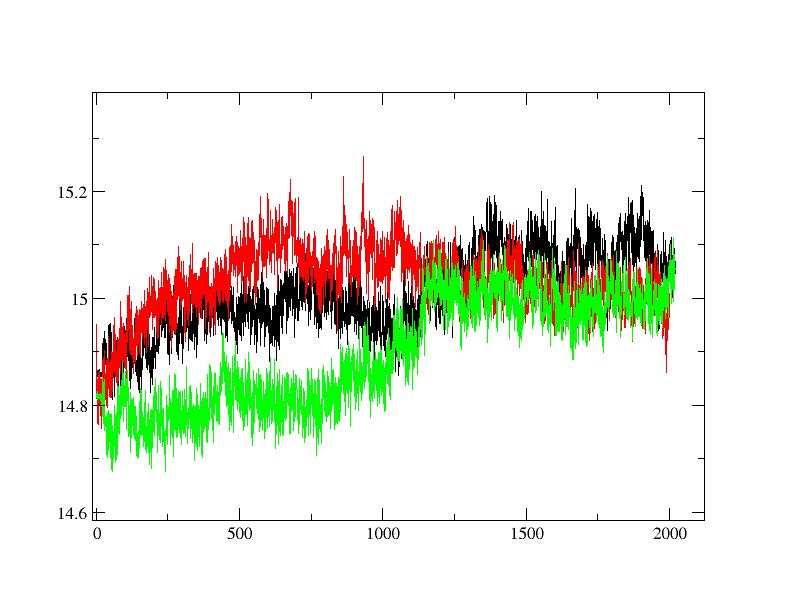
(image/jpeg attachment: RAD-GYR.jpg)
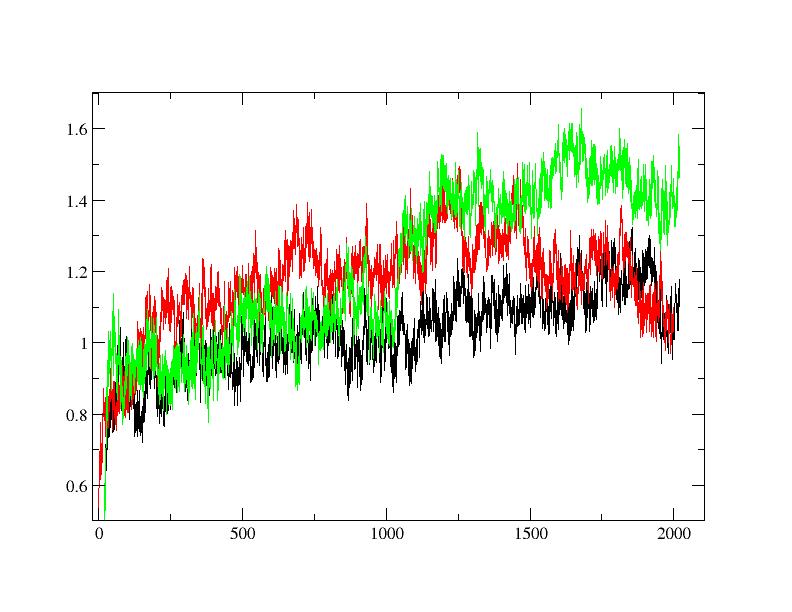
(image/jpeg attachment: rmsd_Ca.jpg)
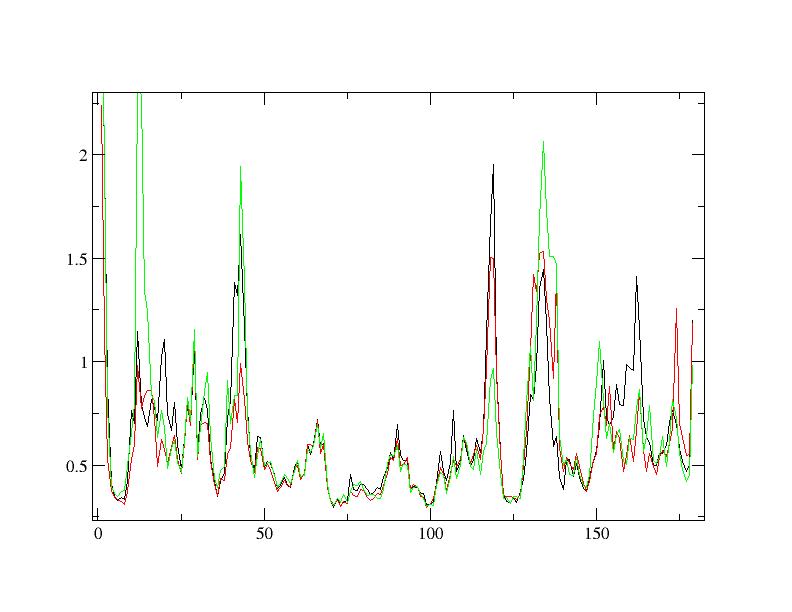
(image/jpeg attachment: rmsf_CA.jpg)
