Dear amber users,
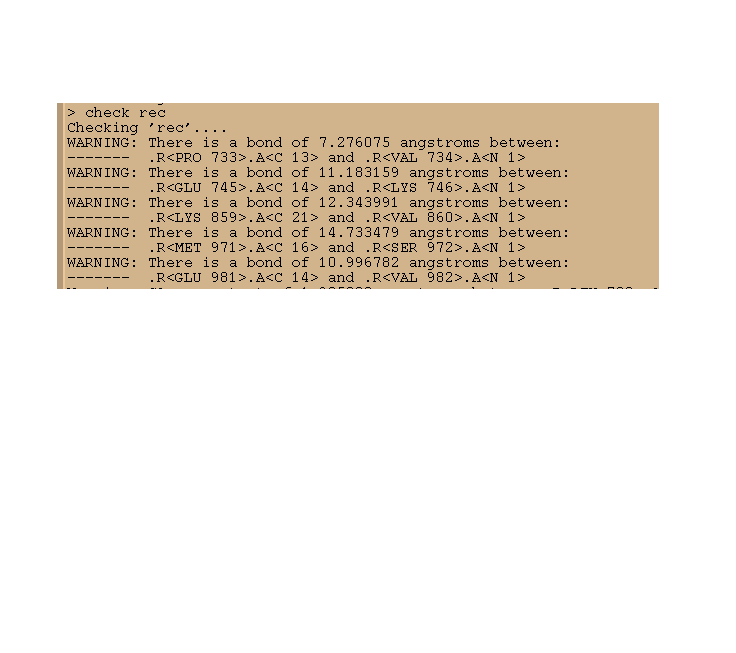
I have a problem when using xleap. when I load the pdb file of receptor, then check it, it gives me those warnings that there are several bonds with a more than 10 angstroms bond length. The attachment file is the pdb file of receptor for you to check for me!
I can't understand that. Could somebody tell me the reason? Any response will be highly appreciated!
All the best!
Qinghua
- x-unknown/x-pdb attachment: rec.pdb
Received on Wed Aug 19 2009 - 19:57:21 PDT
{kind=link}
