Date: Fri, 26 Jun 2009 07:56:35 +0100
Dear Susanne,
> I don't use a script.
I think you should use the scripting approach, add the mode "verbosity
2" & first hunt all the error messages in particular these related to
this Zinc atom.
I created a script for your problem, adding "addAtomTypes": The ZNA
atom types is not possible: Only two carcaters are allowed for FF atom
types.
I modifed your ZNA.lib + zna.frcmod files & generated the prmtop/prmcrd files.
> i will write down, what I do.
> 1) source leaprc.ff03
> 2) source leaprc.gaff
> 3) loadoff ZNA.lib
> 4)loadamberparams zna.frcmod
> 5)loadamberparams bzim_correct.frcmod
> 6) loadamberprep bzim_correct.prepin
> 7)test=loadpdb "c2k_bzim_correct.pdb"
> 8)addions test Cl- 0
> 9) solvateoct test TIP3BOX 8.0
> 10) saveamberparm test test.prmtop test.inpcrd
Below is the script I used:
source leaprc.ff03
source leaprc.gaff
verbosity 2
alias q quit
alias e edit
alias c charge
addAtomTypes {{ "ZN" "Zn""sp3" }}
loadoff ZNA.lib
loadamberparams frcmod.zna
loadamberparams bzim_correct.frcmod
loadamberprep bzim_correct.prepin
test = loadpdb c2k_bzim_correct.pdb
solvateoct test TIP3PBOX 10.0
# add ions after creating the box: this will save you computer time ;-)
addions test Cl- 0
saveamberparm test test.prmtop test.inpcrd
c ABI
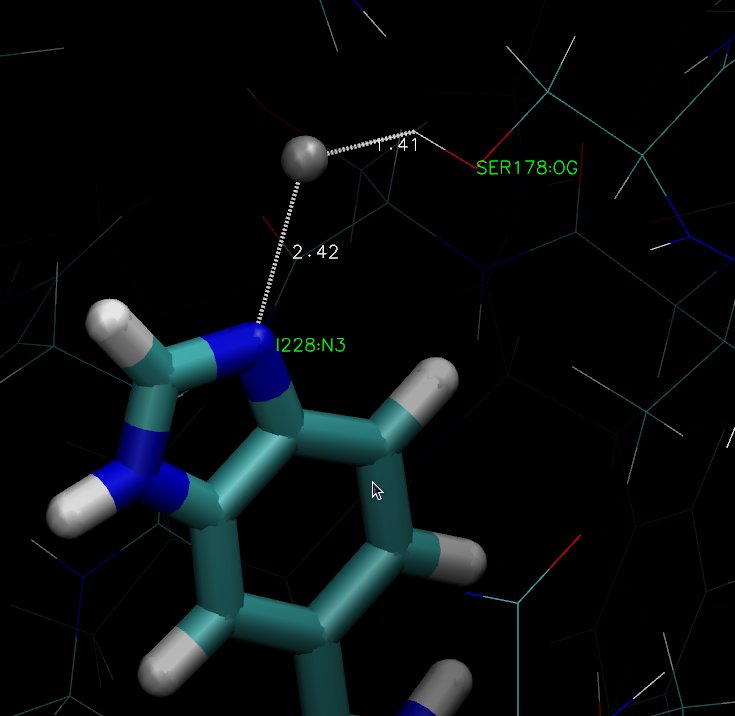
Then, I looked at your protein & saw that you have an hydroxyl (Serine
178; see attached picture ABI.jpg) close to the Zn2+. Does it make
sense ? It should not be an alcoholate ?
During the geometry optimization step I would print the energy values
every step so that you can follow what's going on at the very
beginning of the optimization process. You might try to save the
coordinates more often as well.
regards, Francois
> -----Ursprüngliche Nachricht-----
> Von: amber-bounces.ambermd.org [mailto:amber-bounces.ambermd.org]Im
> Auftrag von FyD
> Gesendet: 25 June 2009 13:45
> An: AMBER Mailing List
> Betreff: Re: [AMBER] atomtypes in gaff
>
>
> Quoting "Aust, Susanne" <saust.ipb-halle.de>:
>
> Could you send me your LEaP script (that used to generate your
> prmtop/prmcrd files) + Zinc library (mol2 file format) + FF library
> for your benzimidazole-structure (mol2 file format) + the structure of
> your complex (PDB file format).
>
> regards, Francois
>
>
>> I load the complex in xleap as pdb, so no name is necessary.
>> At my first trail I load the wrong pdb with ZNB in the pdb. I correct this
> to
>> ZNA (like in my ZNA.lib) and load the pdb again and then everything was
> read
>> in without error.
>> The ligand in the complex has the name ABI. The parameter for this ligand I
>> read in to xleap with loadamberparams bzim_correct.frcmod and loadamberprep
>> bzim_correct.prepin.
>> When I prepare my complex in xleap, there is no error and the pdb or mol2
>> from xleap looks o.k.
>> But after the minimization the ligand is not coordinated. With a wrong
>> benzimidazole-structure (the double bonds are not on the correct positions)
>> the ligand keeps coordinated.
>> It's difficult for me, to describe the problem, because I don't know
> exactly
>> at which position of the preparation line the failure occurs.
>> regards,
>> Susanne
>>
>>
>>
>> -----Ursprüngliche Nachricht-----
>> Von: amber-bounces.ambermd.org [mailto:amber-bounces.ambermd.org]Im
>> Auftrag von FyD
>> Gesendet: 25 June 2009 12:33
>> An: AMBER Mailing List
>> Betreff: Re: AW: [AMBER] atomtypes in gaff
>>
>>
>> Susanne,
>>
>> You need to add a molecule name to your Tripos mol2 file (I wonder if
>> this is not a bug in the savemol2 command because you have many
>> residues...):
>> .<TRIPOS>MOLECULE
>> C2K
>> 3263 3293 228 0 1
>> SMALL
>> etc...
>>
>>> The atomtypes should be correct.
>>> I send you the mol2 file of the comlex from xleap.
>>> The benzimidazole is over the N3 (nd) non physical coordinated to the
> zinc.
>>> I calculate other ligands (substrate and imidazole derivative) with this
>>> enzyme and it works.
>>> So I think it is not a problem of zinc.
>>> I hope, this is the file you want.
>>> I send you also the xleap.log
>>
>> In your LEaP log file, you load the Zinc FF library:
>> loadoff ZNA.lib
>>
>> In your Tripos mol2 file, your Zinc is defined as ZNB (residue name,
>> FF atom type & atom name). Consequently, in LeaP you get this message:
>>
>> Unknown residue: ZNB number: 225 type: Terminal/last
>> ..relaxing end constraints to try for a dbase match
>> -no luck
>> (Residue 226: ZNB, Terminal/last, was not found in name map.)
>> Unknown residue: ZNB number: 226 type: Terminal/last
>> ..relaxing end constraints to try for a dbase match
>> -no luck
>>
>> Then, you load the frcmod file for your zinc atom:
>> Loading parameters: ./frcmod.zna
>> Reading force field modification type file (frcmod)
>> Reading title:
>> This frcmod file is for ZNA (zinc 2+ ion).
>> (UNKNOWN ATOM TYPE: ZNA)
>>
>> I wonder if the FF atom type of Zn is well defined.
>>
>> => add
>> addAtomTypes {
>> { "ZN" "Zn" "sp3" }
>> ^^^^
>> or what is required...
>> }
>>
>> I do not know if these are the reasons of your problem. However, you
>> might correct these problems to see if this helps.
>>
>> regards, Francois
>>
>>
>>
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>>
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>>
>>
>
>
>
> F.-Y. Dupradeau
> ---
> http://q4md-forcefieldtools.org/FyD/
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
F.-Y. Dupradeau
---
http://q4md-forcefieldtools.org/FyD/
(image/jpeg attachment: ABI.jpg)
- text/plain attachment: ATT00002.txt
