Date: Tue, 9 Jun 2009 11:49:43 +0100
Dear amber users,
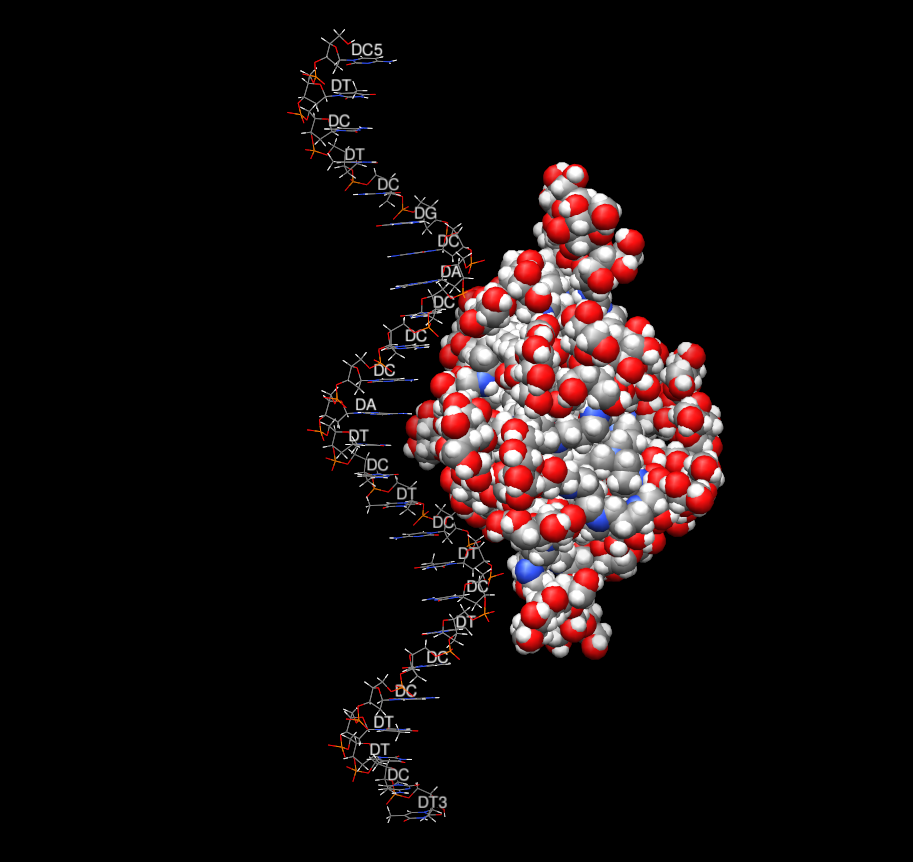
I have done simulation of complex PPI dendrimer - 4th generation which is
decorated with maltose (whole this molecule is electrically neutral)
with single strand DNA (25 bases, with charge -24) in salt explicit water
(ionic strength - 0.15M)- please see the illustrative picture.
The total length of the simulation was 11 ns (4 ns NPT + 7 ns NVT,
T=310K). Below you can find all "in" files.
For the parametrisation I used these forcefields - ff99SB (DNA), gaff
(PPI), glycam (maltose).
Than I made mm_PBSA energetical analysis using the last 2.5 ns (I took 50
frames in 0.1 ns interval).
Here are the final results:
One trajectory approach:
dG = dH - dS = -53.67 - (-74.53) = 20.86
Three trajectories approach:
dG = dH - dS = -15.27 - (-78.67) = 63.4
Maybe it is worth to notice that to speed up the NN analysis I used
separate parallel sander minimisation (conj-grad) using input file
generated
by mm_pbsa routine:
-----------------------------------------------
File generated by mm_pbsa.pl
&cntrl
ntxo = 1,
ntf = 1, ntb = 0,
dielc = 4,
cut = 99.0, nsnb = 99999,
scnb = 2.0, scee = 1.2,
imin = 1, maxcyc = 100000,
ncyc = 0, drms = 0.0001,
&end
&ewald
eedmeth= 5,
&end
----------------------------------------------
I am really not sure how to interpret above reported results, I am not
sure if they are even reliable ...
I would be really grateful for any comments to this strange results.
It is worth to play with some mm_pbsa parameters (to obtain lower dH
contribution and so less positive dG ?)
or should I try to repeat all the analysis with NAB instead of mm_PBSA
routine ?
Thank you very much in advance for any ideas !
With the best regards
Marek
Please find below all the important files/data:
///////THE INPUT FILES FOR MINIMISATION AND
MD////////////////////////////////
#1 - minimisation with restraints
minimise ras-raf
&cntrl
imin=1,
ntmin = 1,
maxcyc=5000,
ncyc=500,
cut=10.0,
ntb=1,
ntpr=100,
ntr=1,
restraintmask=':1-150',
restraint_wt=5.0,
/
#2 - minimistaion without restraints
minimise ras-raf
&cntrl
imin=1,maxcyc=5000,ncyc=2000,
cut=10.0,ntb=1,
ntpr=100,
/
#3 - heating
heat ras-raf
&cntrl
imin=0,irest=1,ntx=5,
nstlim=50000,dt=0.001,
ntc=2,ntf=2,
cut=10.0, ntb=2,
ntp=1,
taup=1.0,
ntpr=5000, ntwx=5000,
ntt=3, gamma_ln=2.0,
temp0=310.0,
ig=-1,
ntr=1, restraintmask=':1-150',
restraint_wt=2.0,
/
#4 - density
heat ras-raf
&cntrl
imin=0,irest=1,ntx=5,
nstlim=250000,dt=0.002,
ntc=2,ntf=2,
cut=10.0, ntb=2, ntp=1, taup=2.0,
ntpr=50000, ntwx=50000,
ntt=3, gamma_ln=2.0,
ig=-1,
temp0=310.0,
/
#5 - 4 ns NPT (equilibrium)
heat ras-raf
&cntrl
imin=0,irest=1,ntx=5,
nstlim=250000,dt=0.002,
ntc=2,ntf=2,
cut=10.0, ntb=2, ntp=1, taup=2.0,
ntpr=50000, ntwx=50000,
ntt=3, gamma_ln=2.0,
ig=-1,
temp0=310.0,
/
#6 - 7 ns NVT (production)
heat ras-raf
&cntrl
imin=0,irest=1,ntx=5,
nstlim=250000,dt=0.002,
ntc=2,ntf=2,
cut=10.0, ntb=1,
ntpr=5000, ntwx=5000,
ntt=3, gamma_ln=2.0,
ig=-1,
temp0=310.0,
/
/////AVERAGES from the last 0.5 ns of simulation
>>> COMPLEX
A V E R A G E S O V E R 50 S T E P S
NSTEP = 250000 TIME(PS) = 11100.000 TEMP(K) = 310.11 PRESS
= 0.0
Etot = -156998.5026 EKtot = 39280.1612 EPtot =
-196278.6638
BOND = 2250.7410 ANGLE = 1813.0047 DIHED =
1060.0015
1-4 NB = 639.7923 1-4 EEL = 7345.4528 VDWAALS =
27593.2769
EELEC = -236980.9330 EHBOND = 0.0000 RESTRAINT =
0.0000
Ewald error estimate: 0.4593E-04
>>> RECEPTOR(dendrimer) - seaparate run
------------------------------------------------------------------------------
A V E R A G E S O V E R 50 S T E P S
NSTEP = 250000 TIME(PS) = 11100.000 TEMP(K) = 309.89 PRESS
= 0.0
Etot = -116491.9660 EKtot = 30052.9725 EPtot =
-146544.9385
BOND = 2031.8582 ANGLE = 1382.5666 DIHED =
496.8121
1-4 NB = 440.5337 1-4 EEL = 10396.5931 VDWAALS =
21710.7831
EELEC = -183004.0852 EHBOND = 0.0000 RESTRAINT =
0.0000
Ewald error estimate: 0.5800E-04
>>> LIGAND(DNA) - separate run
A V E R A G E S O V E R 50 S T E P S
NSTEP = 250000 TIME(PS) = 11100.000 TEMP(K) = 309.94 PRESS
= 0.0
Etot = -91503.2207 EKtot = 20857.8449 EPtot =
-112361.0656
BOND = 206.6247 ANGLE = 447.1949 DIHED =
527.7110
1-4 NB = 213.5335 1-4 EEL = -3008.4689 VDWAALS =
14905.0176
EELEC = -125652.6782 EHBOND = 0.0000 RESTRAINT =
0.0000
Ewald error estimate: 0.6680E-04
//////ENERGETICAL ANALYSIS using mm_PBSA (reported are just files for
single trajectory approach/ in the case
of 3 traj I used the same just with minor changes in )
*******ENTHALPY CONTRIBUTION
*******ENTHALPY CONTRIBUTION
*******ENTHALPY CONTRIBUTION
*******ENTHALPY CONTRIBUTION
#
# Input parameters for mm_pbsa.pl
#
# Holger Gohlke
# 08.01.2002
#
################################################################################
.GENERAL
#
# General parameters
# 0: means NO; >0: means YES
#
# mm_pbsa allows to calculate (absolute) free energies for one molecular
# species or a free energy difference according to:
#
# Receptor + Ligand = Complex,
# DeltaG = G(Complex) - G(Receptor) - G(Ligand).
#
# PREFIX - To the prefix, "{_com, _rec, _lig}.crd.Number" is added during
# generation of snapshots as well as during mm_pbsa
calculations.
# PATH - Specifies the location where to store or get snapshots.
#
# COMPLEX - Set to 1 if free energy difference is calculated.
# RECEPTOR - Set to 1 if either (absolute) free energy or free energy
# difference are calculated.
# LIGAND - Set to 1 if free energy difference is calculated.
#
# COMPT - parmtop file for the complex (not necessary for option GC).
# RECPT - parmtop file for the receptor (not necessary for option GC).
# LIGPT - parmtop file for the ligand (not necessary for option GC).
#
# GC - Snapshots are generated from trajectories (see below).
# AS - Residues are mutated during generation of snapshots from
trajectories.
# DC - Decompose the free energies into individual contributions
# (only works with MM and GB).
#
# MM - Calculation of gas phase energies using sander.
# GB - Calculation of desolvation free energies using the GB models in
sander
# (see below).
# PB - Calculation of desolvation free energies using delphi (see below).
# Calculation of nonpolar solvation free energies according to
# the NPOPT option in pbsa (see below).
# MS - Calculation of nonpolar contributions to desolvation using molsurf
# (see below).
# If MS == 0 and GB == 1, nonpolar contributions are calculated
with the
# LCPO method in sander.
# If MS == 0 and PB == 1, nonpolar contributions are calculated
according
# the NPOPT option in pbsa (see below).
# NM - Calculation of entropies with nmode.
#
PREFIX snap
PATH ./
#
COMPLEX 1
RECEPTOR 1
LIGAND 1
#
COMPT ./com.prmtop
RECPT ./rec.prmtop
LIGPT ./lig.prmtop
#
GC 0
AS 0
DC 0
#
MM 1
GB 0
PB 1
MS 1
#
NM 0
#
################################################################################
.PB
#
# PB parameters (this section is only relevant if PB = 1 above)
#
# The following parameters are passed to the PB solver.
# Additional input parameters may also be added here. See the sander PB
# documentation for more options.
#
# PROC - Determines which method is used for solving the PB equation:
# By default, PROC = 2, the pbsa program of the AMBER suite is
used.
# REFE - Determines which reference state is taken for PB calc:
# By default, REFE = 0, reaction field energy is calculated with
# EXDI/INDI. Here, INDI must agree with DIELC from MM part.
# INDI - Dielectric constant for the solute.
# EXDI - Dielectric constant for the surrounding solvent.
# ISTRNG - Ionic strength (in mM) for the Poisson-Boltzmann solvent.
# PRBRAD - Solvent probe radius in Angstrom:
# 1.4: with the radii in the prmtop files. Default.
# 1.6: with the radii optimized by Tan and Luo (In preparation).
# See RADIOPT on how to choose a cavity radii set.
# RADIOPT - Option to set up radii for PB calc:
# 0: uses the radii from the prmtop file. Default.
# 1: uses the radii optimized by Tan and Luo (In preparation)
# with respect to the reaction field energies computed
# in the TIP3P explicit solvents. Note that optimized radii
# are based on AMBER atom types (upper case) and charges.
# Radii from the prmtop files are used if the atom types
# are defined by antechamber (lower case).
# SCALE - Lattice spacing in no. of grids per Angstrom.
# LINIT - No. of iterations with linear PB equation.
#
# NP Parameters for nonpolar solvation energies if MS = 0
#
# NPOPT - Option for modeling nonpolar solvation free energy.
# See sander PB documentation for more information on the
# implementations by Tan and Luo (In preparation).
# 1: uses the solvent-accessible-surface area to correlate total
# nonpolar solvation free energy:
# Gnp = CAVITY_SURFTEN * SASA + CAVITY_OFFSET. Default.
# 2: uses the solvent-accessible-surface area to correlate the
# repulsive (cavity) term only, and uses a surface-integration
# approach to compute the attractive (dispersion) term:
# Gnp = Gdisp + Gcavity
# = Gdisp + CAVITY_SURFTEN * SASA + CAVITY_OFFSET.
# When this option is used, RADIOPT has to be set to 1,
# i.e. the radii set optimized by Tan and Luo to mimic Gnp
# in TIP3P explicit solvents. Otherwise, there is no guarantee
# that Gnp matches that in explicit solvents.
# CAVITY_SURFTEN/CAVITY_OFFSET - Values used to compute the nonpolar
# solvation free energy Gnp according NPOPT. The default values
# are for NPOPT set to 0 and RADIOPT set to 0 (see above).
# If NPOPT is set to 1 and RADIOPT set to 1, these two lines
# can be removed, i.e. use the default values set in pbsa
# for this nonpolar solvation model. Otherwise, please
# set these to the following:
# CAVITY_SURFTEN: 0.04356
# CAVITY_OFFSET: -1.008
#
# NP Parameters for nonpolar solvation energies if MS = 1
#
# SURFTEN/SURFOFF - Values used to compute the nonpolar contribution Gnp
to
# the desolvation according to Gnp = SURFTEN * SASA + SURFOFF.
#
PROC 2
REFE 0
INDI 1.0
EXDI 80.0
SCALE 2
LINIT 1000
PRBRAD 1.4
ISTRNG 0.0
RADIOPT 0
NPOPT 1
CAVITY_SURFTEN 0.0072
CAVITY_OFFSET 0.00
#
SURFTEN 0.0072
SURFOFF 0.00
#
################################################################################
.MM
#
# MM parameters (this section is only relevant if MM = 1 above)
#
# The following parameters are passed to sander.
# For further details see the sander documentation.
#
# DIELC - Dielectricity constant for electrostatic interactions.
# Note: This is not related to GB calculations.
#
DIELC 1.0
#
################################################################################
.GB
#
# GB parameters (this section is only relevant if GB = 1 above)
#
# The first group of the following parameters are passed to sander.
# For further details see the sander documentation.
#
# IGB - Switches between Tsui's GB (1), Onufriev's GB (2, 5).
# GBSA - Switches between LCPO (1) and ICOSA (2) method for SASA calc.
# Decomposition only works with ICOSA.
# SALTCON - Concentration (in M) of 1-1 mobile counterions in solution.
# EXTDIEL - Dielectricity constant for the solvent.
# INTDIEL - Dielectricity constant for the solute
#
# SURFTEN / SURFOFF - Values used to compute the nonpolar contribution
Gnp to
# the desolvation according to Gnp = SURFTEN * SASA +
SURFOFF.
#
IGB 2
GBSA 1
SALTCON 0.00
EXTDIEL 80.0
INTDIEL 1.0
#
SURFTEN 0.0072
SURFOFF 0.00
#
################################################################################
.MS
#
# Molsurf parameters (this section is only relevant if MS = 1 above)
#
# PROBE - Radius of the probe sphere used to calculate the SAS.
# Since Bondi radii are already augmented by 1.4A, PROBE should
be 0.0
#
PROBE 0.0
#
#################################################################################
.PROGRAMS
#
# Additional program executables can be defined here
#
#
################################################################################
************INPUT FILE for NN analysis
************INPUT FILE for NN analysis
************INPUT FILE for NN analysis
************INPUT FILE for NN analysis
#
# Input parameters for mm_pbsa.pl
#
# Holger Gohlke
# 08.01.2002
#
################################################################################
.GENERAL
#
# General parameters
# 0: means NO; >0: means YES
#
# mm_pbsa allows to calculate (absolute) free energies for one molecular
# species or a free energy difference according to:
#
# Receptor + Ligand = Complex,
# DeltaG = G(Complex) - G(Receptor) - G(Ligand).
#
# PREFIX - To the prefix, "{_com, _rec, _lig}.crd.Number" is added during
# generation of snapshots as well as during mm_pbsa
calculations.
# PATH - Specifies the location where to store or get snapshots.
#
# COMPLEX - Set to 1 if free energy difference is calculated.
# RECEPTOR - Set to 1 if either (absolute) free energy or free energy
# difference are calculated.
# LIGAND - Set to 1 if free energy difference is calculated.
#
# COMPT - parmtop file for the complex (not necessary for option GC).
# RECPT - parmtop file for the receptor (not necessary for option GC).
# LIGPT - parmtop file for the ligand (not necessary for option GC).
#
# GC - Snapshots are generated from trajectories (see below).
# AS - Residues are mutated during generation of snapshots from
trajectories.
# DC - Decompose the free energies into individual contributions
# (only works with MM and GB).
#
# MM - Calculation of gas phase energies using sander.
# GB - Calculation of desolvation free energies using the GB models in
sander
# (see below).
# PB - Calculation of desolvation free energies using delphi (see below).
# Calculation of nonpolar solvation free energies according to
# the NPOPT option in pbsa (see below).
# MS - Calculation of nonpolar contributions to desolvation using molsurf
# (see below).
# If MS == 0 and GB == 1, nonpolar contributions are calculated
with the
# LCPO method in sander.
# If MS == 0 and PB == 1, nonpolar contributions are calculated
according
# the NPOPT option in pbsa (see below).
# NM - Calculation of entropies with nmode.
#
PREFIX snap
PATH ./
#
COMPLEX 1
RECEPTOR 1
LIGAND 1
#
COMPT ./com.prmtop
RECPT ./rec.prmtop
LIGPT ./lig.prmtop
#
GC 0
AS 0
DC 0
#
MM 0
GB 0
PB 0
MS 0
#
NM 1
#
#
#################################################################################
.NM
#
# Parameters for sander/nmode calculation (this section is only relevant
# if NM = 1 above)
#
# The following parameters are passed to sander (for minimization) and
nmode
# (for entropy calculation using gasphase statistical mechanics).
# For further details see documentation.
#
# DIELC - (Distance-dependent) dielectric constant
# MAXCYC - Maximum number of cycles of minimization.
# DRMS - Convergence criterion for the energy gradient.
#
DIELC 4
MAXCYC 30
DRMS 0.0001
#
#################################################################################.PROGRAMS
#
# Additional program executables can be defined here
#
#
################################################################################
-- Tato zpráva byla vytvořena převratným poštovním klientem Opery: http://www.opera.com/mail/
(image/png attachment: PPI4_malt_ssDNA.png)
- text/plain attachment: ATT00002.txt
