Date: Thu, 8 Jan 2009 10:25:57 -0700
Hello,
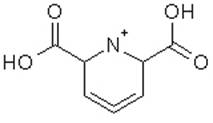
We would like to run molecular dynamics simulations for the compound shown below. Is Amber the correct force field for this? Or are there other more appropriate force fields?
[cid:image001.jpg.01C9717B.7E957550]
--------------------------------------------
Richard
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: image001.jpg)
