Date: Wed, 10 Dec 2008 12:33:27 +0900
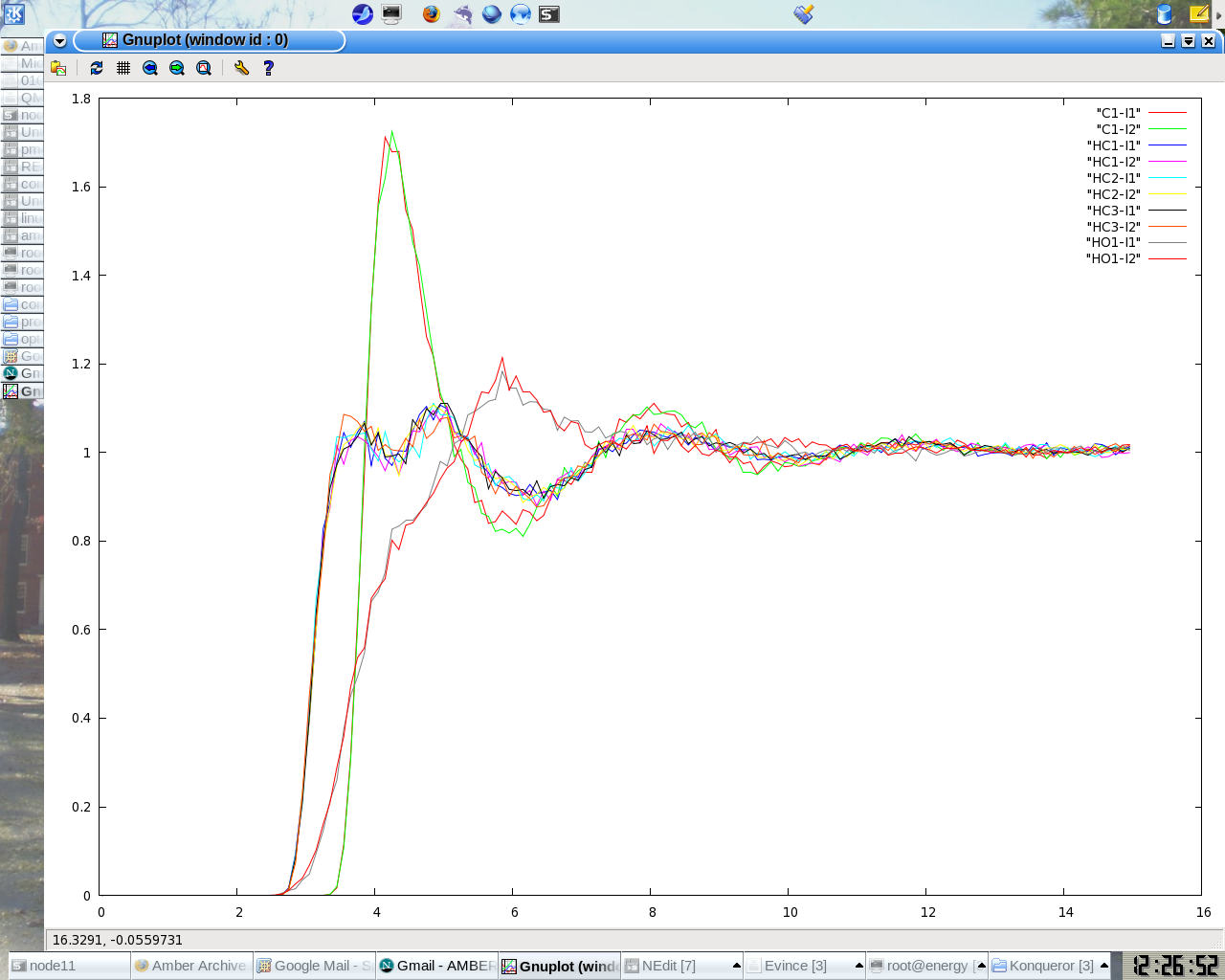
Thank you Dr.Gustavo. I checked and corrected the names of the hydrogen
verifying it from leap and got it fixed. now all the RDFs pairs are
converging to 1. But I find that the RDF curve is very noisy( am saving the
coordinate every 200fs and the time step is 0.5fs), when compared to that of
moldy simulations, any suggestions.?
regards
prabhakar
On Wed, Dec 10, 2008 at 12:38 AM, Gustavo Seabra
<gustavo.seabra.gmail.com>wrote:
> Hi there,
>
> Could you check your ptraj output for the results of the atom
> selection masks you are using? That is output in the begginign og the
> ptraj output file.
>
> The plot you show only has 3 distinctive sets of lines: The first two
> come from the C-I and O-I RDFs, and the last set is uncertain, but
> probably bunches together everything that contain Hydrogens. My guess
> is that ptraj is not completely understanding your selection masks
> when it comes to the Hydrogens.
>
> Gustavo.
>
> On Mon, Dec 8, 2008 at 9:37 PM, prabhakar g wrote:
> > Dear Amber experts,
> >
> > I have carried out a 1ns restrained md simulation of I2(iodide) molecule
> in
> > MEOHBOX using ff99 force field,
> > (force constant of 100 kcal/mol)
> > Here is my simulation protocol using sander(Amber9)(from tutorial)
> > Minimization I (restrained minimization)
> > Minimization II ( whole system minimization)
> > Heating 20ps(NVT)
> > equilibration 300ps (NPT)
> > production 1ns (NVT)
> >
> > am interested in calculating the radial distribution function(rdf) for
> > studies related to photo-dissociation.
> > I calculated the rdf with ptraj but for some of the atomic pairs i am
> > getting an unusual rdf
> > that is the curve does not converges to 1 for some of the atomic pairs (
> > file attached)
> > here is my ptraj input
> >
> > trajin i2_meoh_md1.2ns.mdcrd
> > radial rdfC1-I1.out 0.1 15.0 :2-569.C1 :1.I1 density 0.015036
> > radial rdfC1-I2.out 0.1 15.0 :2-569.C1 :1.I2 density 0.015036
> > radial rdfO1-I1.out 0.1 15.0 :2-569.O1 :1.I1 density 0.015036
> > radial rdfO1-I2.out 0.1 15.0 :2-569.O1 :1.I2 density 0.015036
> > radial rdf1HC-I1.out 0.1 15.0 :2-569.1HC :1.I1 density 0.015036
> > radial rdf1HC-I2.out 0.1 15.0 :2-569.1HC :1.I2 density 0.015036
> > radial rdf2HC-I1.out 0.1 15.0 :2-569.2HC :1.I1 density 0.015036
> > radial rdf2HC-I2.out 0.1 15.0 :2-569.2HC :1.I2 density 0.015036
> > radial rdf3HC-I1.out 0.1 15.0 :2-569.3HC :1.I1 density 0.015036
> > radial rdf3HC-I2.out 0.1 15.0 :2-569.3HC :1.I2 density 0.015036
> > radial rdf1HO-I1.out 0.1 15.0 :2-569.1HO :1.I1 density 0.015036
> > radial rdf1HO-I2.out 0.1 15.0 :2-569.1HO :1.I2 density 0.015036
> >
> > whereas the same simulation in moldy(
> http://www.ccp5.ac.uk/moldy/moldy.html)
> > I get a very gud radial distrubution function. my aim was to scale the
> > computing time of both the programs ..
> > could anyone help regarding this..
> >
> > thanks in advance
> > prabhakar
> >
>
>
>
> --
> Gustavo Seabra
> Postdoctoral Associate
> Quantum Theory Project - University of Florida
> Gainesville - Florida - USA
> -----------------------------------------------------------------------
> The AMBER Mail Reflector
> To post, send mail to amber.scripps.edu
> To unsubscribe, send "unsubscribe amber" (in the *body* of the email)
> to majordomo.scripps.edu
>
-----------------------------------------------------------------------
The AMBER Mail Reflector
To post, send mail to amber.scripps.edu
To unsubscribe, send "unsubscribe amber" (in the *body* of the email)
to majordomo.scripps.edu
(image/png attachment: rdf1.png)
