Date: Tue, 9 Dec 2008 11:37:52 +0900
Dear Amber experts,
I have carried out a 1ns restrained md simulation of I2(iodide) molecule in
MEOHBOX using ff99 force field,
(force constant of 100 kcal/mol)
Here is my simulation protocol using sander(Amber9)(from tutorial)
Minimization I (restrained minimization)
Minimization II ( whole system minimization)
Heating 20ps(NVT)
equilibration 300ps (NPT)
production 1ns (NVT)
am interested in calculating the radial distribution function(rdf) for
studies related to photo-dissociation.
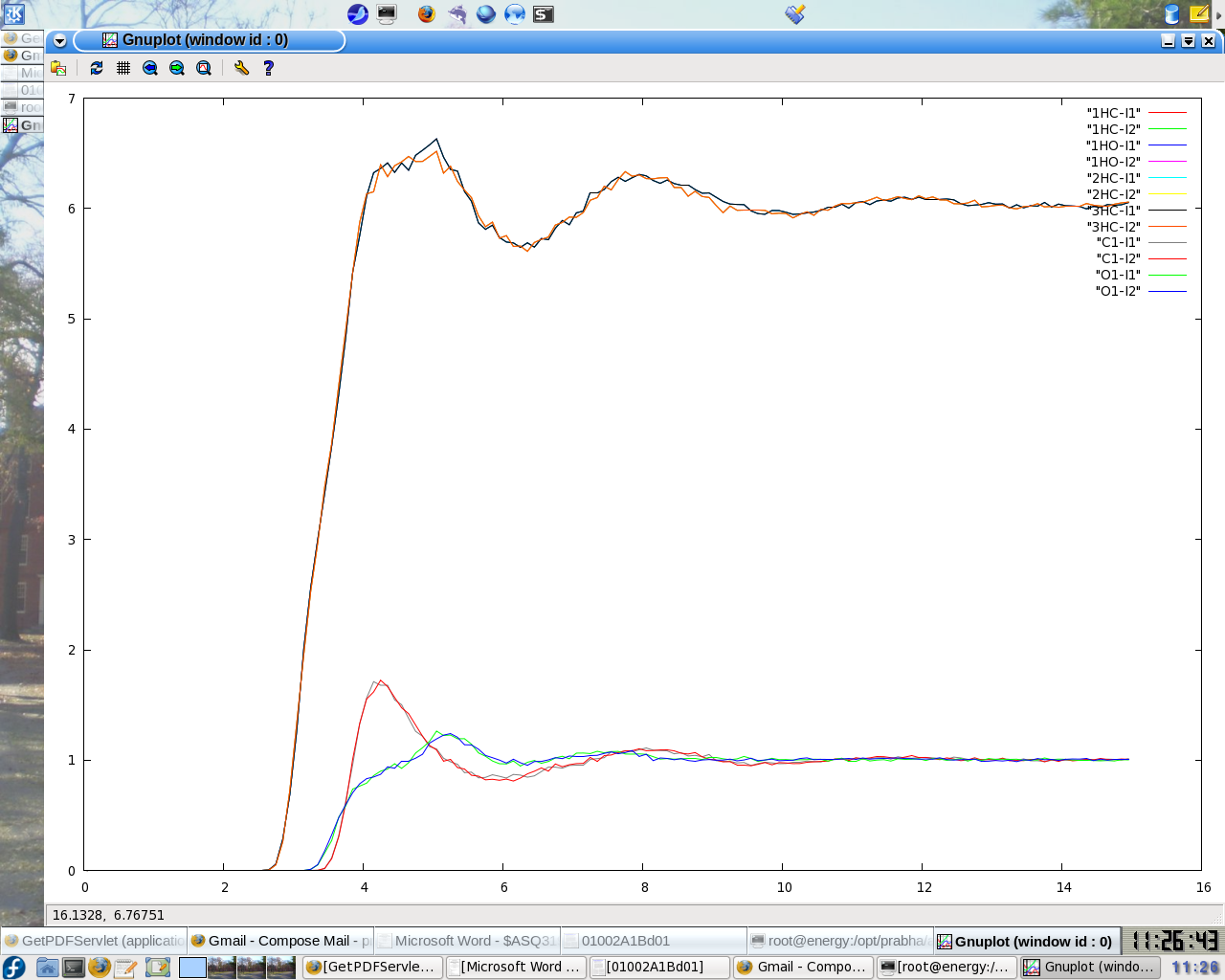
I calculated the rdf with ptraj but for some of the atomic pairs i am
getting an unusual rdf
that is the curve does not converges to 1 for some of the atomic pairs (
file attached)
here is my ptraj input
trajin i2_meoh_md1.2ns.mdcrd
radial rdfC1-I1.out 0.1 15.0 :2-569.C1 :1.I1 density 0.015036
radial rdfC1-I2.out 0.1 15.0 :2-569.C1 :1.I2 density 0.015036
radial rdfO1-I1.out 0.1 15.0 :2-569.O1 :1.I1 density 0.015036
radial rdfO1-I2.out 0.1 15.0 :2-569.O1 :1.I2 density 0.015036
radial rdf1HC-I1.out 0.1 15.0 :2-569.1HC :1.I1 density 0.015036
radial rdf1HC-I2.out 0.1 15.0 :2-569.1HC :1.I2 density 0.015036
radial rdf2HC-I1.out 0.1 15.0 :2-569.2HC :1.I1 density 0.015036
radial rdf2HC-I2.out 0.1 15.0 :2-569.2HC :1.I2 density 0.015036
radial rdf3HC-I1.out 0.1 15.0 :2-569.3HC :1.I1 density 0.015036
radial rdf3HC-I2.out 0.1 15.0 :2-569.3HC :1.I2 density 0.015036
radial rdf1HO-I1.out 0.1 15.0 :2-569.1HO :1.I1 density 0.015036
radial rdf1HO-I2.out 0.1 15.0 :2-569.1HO :1.I2 density 0.015036
whereas the same simulation in moldy(http://www.ccp5.ac.uk/moldy/moldy.html)
I get a very gud radial distrubution function. my aim was to scale the
computing time of both the programs ..
could anyone help regarding this..
thanks in advance
prabhakar
-----------------------------------------------------------------------
The AMBER Mail Reflector
To post, send mail to amber.scripps.edu
To unsubscribe, send "unsubscribe amber" (in the *body* of the email)
to majordomo.scripps.edu
(image/png attachment: rdf.png)
