Date: Fri, 4 Jun 2004 17:21:20 +0800
Dear Amber users,
Sorry for that this letter is relatively long.
I encountered a strange problem in my GBSA run. I found
the potential and total energy of the native state
of protein CI2 is even higher than that of non-native
state. But this is not correct.
I made two runs at temperature 100K, one was started from
the NMR structure of the native state of protein CI2, the
other was started from a relative random structure (see
the attached pdb files). Minimizations have been made before
the MD runs. In the calculations, the ff99 force field
and AMBER7 is used.
The mdin files in two runs are the same:
------------------------------------------
constant temperature GBSA run
&cntrl
imin=0, ntb=0, ntt=1, ntc=2, ntf=2, dt=0.002, cut=8.0,
igb=2, gbsa=1,
ntpr = 10, ntwr=10, ntwx=10, ntwe=10,
nstlim = 1000000,
temp0 = 100,
tempi = 100,
tautp = 0.1,
&end
END
------------------------------------------
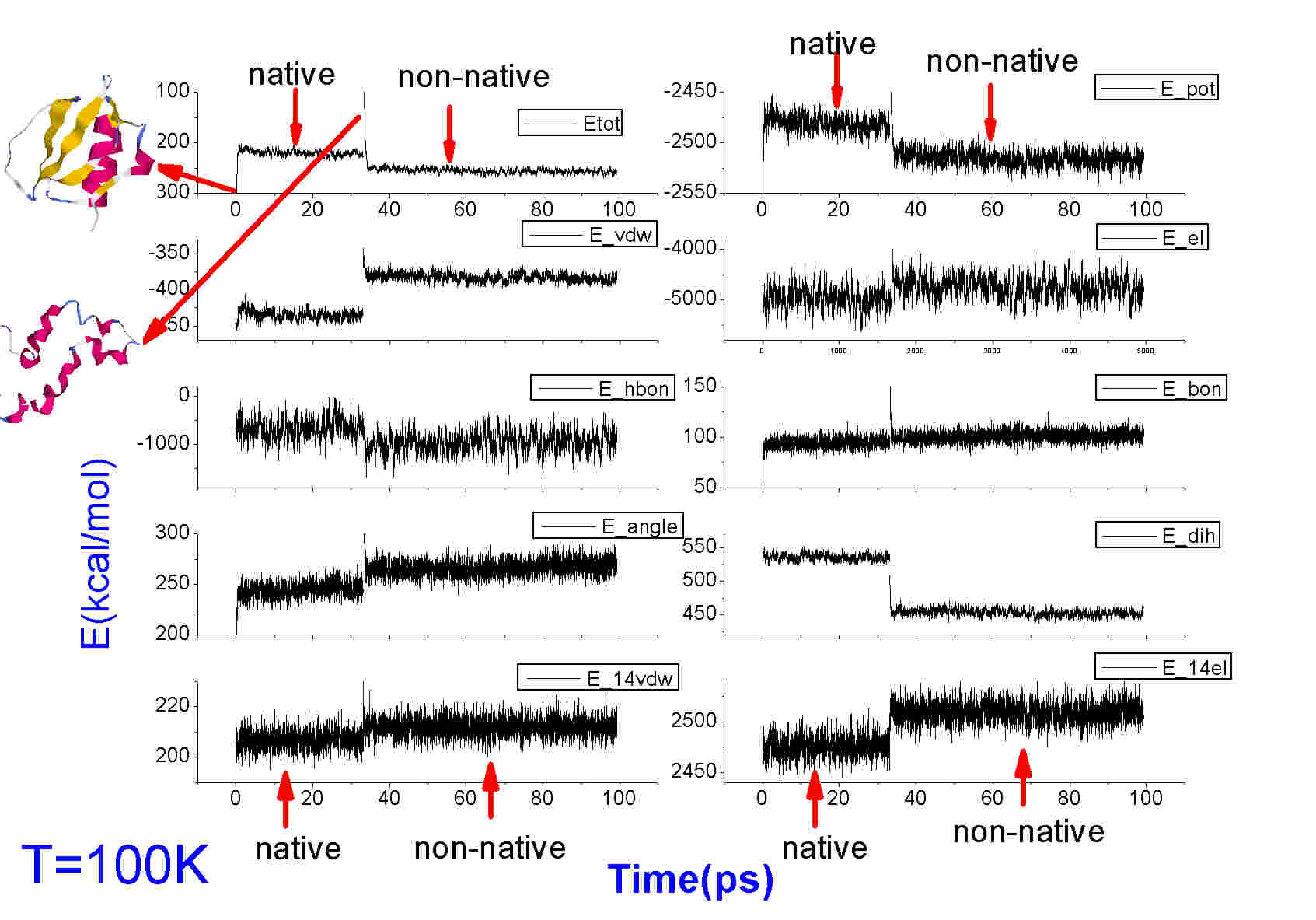
The obtained energies (from mden files) are given in
the attached JPG file. To compare the two trajectories,
I put the energies of two trajectories together in
each figure. Long time simulations have been made to
ensure equilibrium, but here I only show the results
of a short time.
I can be seen that the potential and total energy of
native state are even higher that that of non-native state!
But this is not correct. The starting structures of two
runs are compared to the finial structures, and they are
almost the same. That is, the structures did not change much.
Similar results have also been seen in temperature 300K.
Why? Is this due to some incorrect parameters in my mdin file,
or due to improper force field chosen? or sth else?
Thank you very much for your help.
Best Regards,
--- J. Zhang, Dr, Institute of Biophysics, Nanjing University
-----------------------------------------------------------------------
The AMBER Mail Reflector
To post, send mail to amber.scripps.edu
To unsubscribe, send "unsubscribe amber" to majordomo.scripps.edu
(image/jpeg attachment: strangEnergy.JPG)
- application/octet-stream attachment: ci2-native.pdb
- application/octet-stream attachment: ci2-non-native.pdb
