Date: Sun, 13 Jul 2025 11:57:11 +0530
Dear Amber community,
Hope this email finds you well. I am Arka Acharyya, a PhD scholar from IIT
Indore, India, Department of Chemistry. I am trying to build the force
parameter for boron in 2-Formylphenylboronic acid by paramfit programme in
AmberTools22. Initially, I built the molecule using GaussView 6.0. Then,
Geometry was optimized using b3lyp/6-31g(d). Then I tried to build a mol2
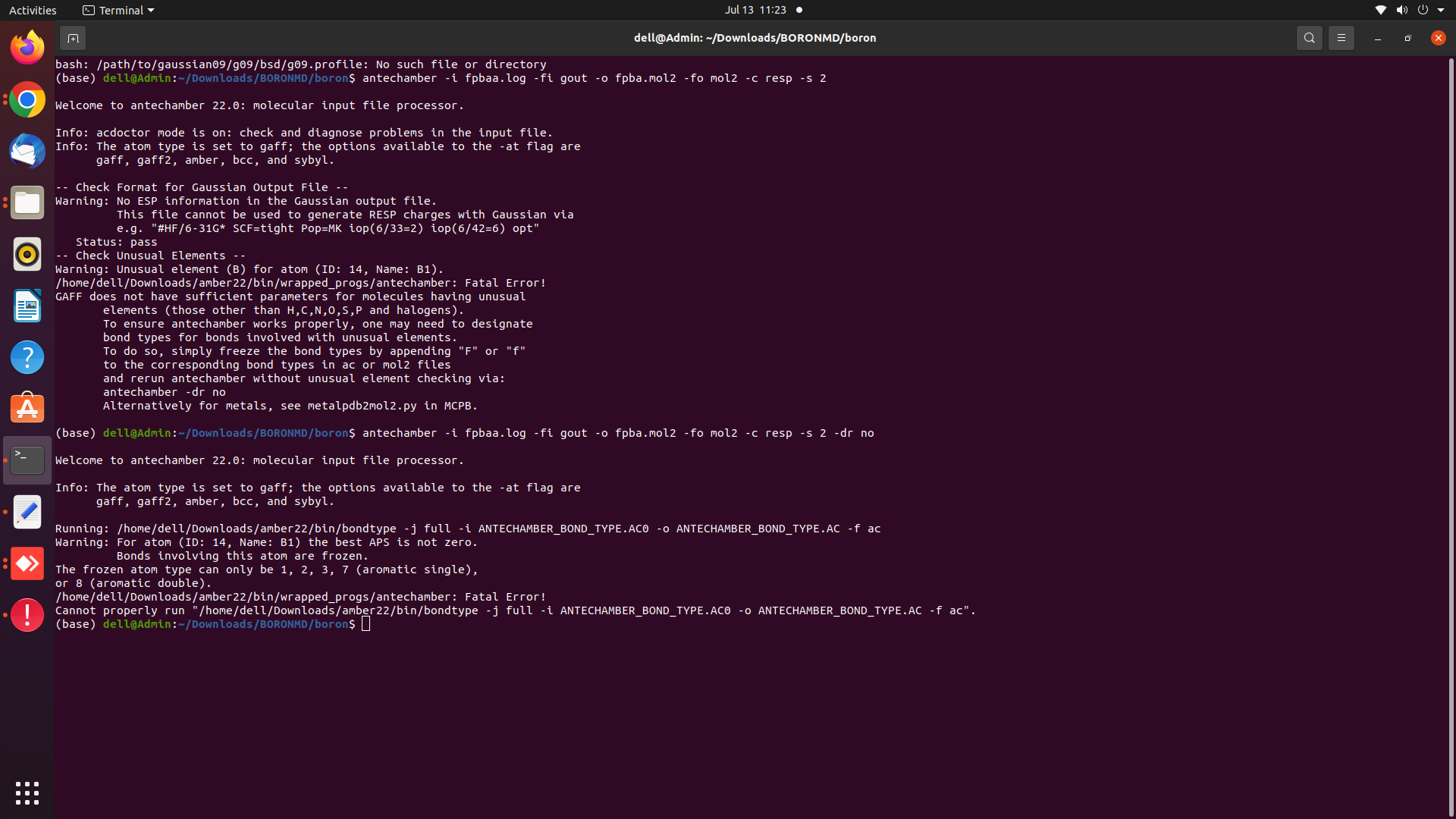
file using antechamber -i fpbaa.log -fi gout -o fpba.mol2 -fo mol2 -c resp
-s 2 -dr no. Then, I encountered the following error
Warning: For atom (ID: 14, Name: B1) the best APS is not zero.
Bonds involving this atom are frozen.
The frozen atom type can only be 1, 2, 3, 7 (aromatic single),
or 8 (aromatic double).
/home/dell/Downloads/amber22/bin/wrapped_progs/antechamber: Fatal Error!
Cannot properly run "/home/dell/Downloads/amber22/bin/bondtype -j full -i
ANTECHAMBER_BOND_TYPE.AC0 -o ANTECHAMBER_BOND_TYPE.AC
<http://antechamber_bond_type.ac/> -f ac".
I am also attaching the screenshot for your reference. Kindly, guide me
with the procedure.
Thank you for your anticipation.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screenshot.png)
