Date: Sat, 22 Feb 2025 04:52:07 +0000
Hello
We have tried running a short simulation of AMBER tutorial DNA polyA-polyT to check our simulations. So according to the tutorial the first run (MD1) was for 20ps and second run (MD2) for 100ps.
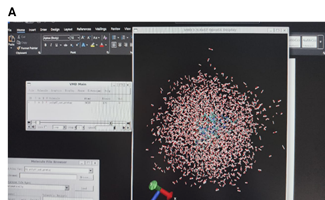
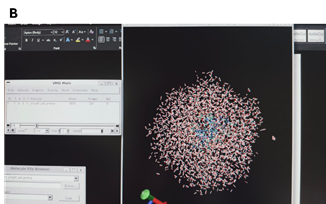
The water box was intact till first 20ps and starts breaking after 20ps as can be seen in images A and B.
[cid:7fb3649b-9f85-4d4c-a802-58ade671376d]
[cid:5874adda-7446-4c3f-a905-3627428be628]
We have also tried visualization with and without prior autoimage and unwrap but the system was still seen broke
________________________________
From: Dr. Anselm Horn via AMBER <amber.ambermd.org>
Sent: 18 February 2025 16:30
To: amber.ambermd.org <amber.ambermd.org>
Subject: Re: [AMBER] Issue with Water Box Breaking in CUDA MD Simulations
Tushar,
did you visualize your trajectory ("watch the movie")? Are your problems
occuring instantaneously from one step to another or building up slowly?
Did you perform short sample runs printing out every step, as Prof. Case
suggested? Is your restraint definition input correct (you state "0.5
kcal/mol" in the title, but define 40 kcal/mol/A**2 in the input) and
also recognized correctly (mirrored in the output file)? Did you also
try visualization without prior autoimage and unwrap? Maybe you did
unwrap only your protein residues?
Good luck.
Anselm
Bioinformatik | NHR.FAU
Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU)
Germany
Am 17.02.2025 um 06:48 schrieb TUSHAR GUPTA via AMBER:
> Thank you for your reply.
> My work includes working on the protein aptamer complex where i am working on the 3HSB protein complex.
> After addition of solvate box and minimization and heating the complex appears normal .as in figure 1, after running equilibration, at 1250000 steps, the water box breaks as well as the protein chains get distorted as in figure 2 and 3.
> I have used autoimage and unwrap command but the issue doesn't seem to resolve.
>
> I am using running the gpu cuda11.7
> Below is the equilibration.in file I am using
>
>
> &cntrl
> imin=0, ntx=7, ntpr=500, ntwr=500, ntwx=500, ntwe=500,
> nscm=5000,
> ntf=2, ntc=2,
> ntb=2, ntp=1, tautp=0.2, taup=0.2,
> nstlim=1250000, t=0.0, dt=0.002,
> cut=9.0,
> ntt=1,
> ntr=1,
> irest=1
> &end
> Group input for restraints: 0.5 kcal/mol.
> 40
> RES 1 411
> END
> END
>
> Please provide your valuable feedback and suggestion for solution
>
> [cid:77cf0154-9954-4cac-ad29-5486c66e8a6d]
> Figure 1
> [cid:f6fcf3c0-1dee-42ad-9e5a-3de0ecdd08a5]
> Figure 2
> [cid:d14ba297-99c4-4eeb-97e8-b82ec15321f2]
> Figure 3
>
> ________________________________
> From: David A Case <dacase1.gmail.com>
> Sent: 13 February 2025 20:11
> To: TUSHAR GUPTA <E24SOEP0001.bennett.edu.in>
> Subject: Re: [AMBER] Issue with Water Box Breaking in CUDA MD Simulations
>
> On Thu, Feb 13, 2025, TUSHAR GUPTA wrote:
>>
>> I have run md1 and md2. md1 for 20ps and md2 for 100ps. After 20ps the
>> water box starts to break. And even using the reimaging command the box
>> still at the same condition.
>
> Were you able to run the short CPU vs GPU tests that I suggested?
>
>> Basically, you are getting different results from CPU and GPU. To start
>> debugging, set up a short simulation (say 20 steps, with ntpr=1) and run
>> twice, once with pmemd (not sander) and again with pmemd.cuda_DPFP. Look
>> carefully at the two outputs to see what you can find.
>
> The hope is that these short runs may help identify the reason for the
> difference between pmemd and pmemd.cuda. I'm guessing that we are going to
> need to see your files, but you should do this test yourself first.
>
> ....dac
>
> “Your attitude, not your aptitude, will determine your altitude.” – Zig Ziglar
>
> _____________________________________________
>
> Vineet Jain (Chancellor) Inspiration
>
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> https://ind01.safelinks.protection.outlook.com/?url=http%3A%2F%2Flists.ambermd.org%2Fmailman%2Flistinfo%2Famber&data=05%7C02%7C%7C423874951f9440fe719608dd500bf87b%7C2c5bdaf48ff24bd9bd547c50ab219590%7C0%7C0%7C638754734565274498%7CUnknown%7CTWFpbGZsb3d8eyJFbXB0eU1hcGkiOnRydWUsIlYiOiIwLjAuMDAwMCIsIlAiOiJXaW4zMiIsIkFOIjoiTWFpbCIsIldUIjoyfQ%3D%3D%7C0%7C%7C%7C&sdata=E8XtryHfQOjx%2FqKlttkzbjVkAiOxj3KmA4cDonY%2F7oo%3D&reserved=0<http://lists.ambermd.org/mailman/listinfo/amber>
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
https://ind01.safelinks.protection.outlook.com/?url=http%3A%2F%2Flists.ambermd.org%2Fmailman%2Flistinfo%2Famber&data=05%7C02%7C%7C423874951f9440fe719608dd500bf87b%7C2c5bdaf48ff24bd9bd547c50ab219590%7C0%7C0%7C638754734565289607%7CUnknown%7CTWFpbGZsb3d8eyJFbXB0eU1hcGkiOnRydWUsIlYiOiIwLjAuMDAwMCIsIlAiOiJXaW4zMiIsIkFOIjoiTWFpbCIsIldUIjoyfQ%3D%3D%7C0%7C%7C%7C&sdata=bB4n2xktsLpZU2zy3jsTVwVR3jypitU%2FydjetU5RmnE%3D&reserved=0<http://lists.ambermd.org/mailman/listinfo/amber>
“If you really want to do something, you’ll find a way. If you don’t, you’ll find an excuse.” – Jim Rohn
_____________________________________________
Vineet Jain (Chancellor) Inspiration
“It is not in the stars to hold our destiny but in ourselves.” – William Shakespeare
_____________________________________________
Vineet Jain (Chancellor) Inspiration
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
(image/png attachment: 02-image.png)
