Date: Mon, 17 Feb 2025 18:14:47 +0900
Dear all,
Hi, I have been trying to calculate the pressure tensor using amber data. I
have stored the force, velocity, and trajectory for each snapshot. Then I
tried to calculate manually in Python using the Green Kubo Equation. As far
as I know ''mden'' file stores the diagonal pressure tensor as Pres_x,
Pres_y and Pres_z in line number 3, I wanted to compare with that but there
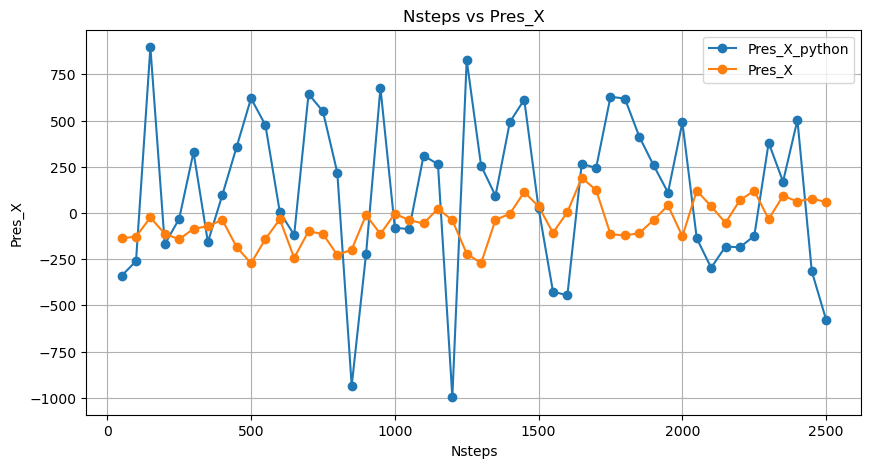
was no correlation with my result(see the graph Nsteps vs Pres_X). To
calculate the pressure tensor, earlier I translated the trajectory in the
box to maintain the boundary condition. However, my final goal is to
calculate the viscosity and I am simulating an ionic liquid of 25000 atoms
with PBC(90*90*90). Can anyone help me with any hints or resources to meet
the queries below:
1. Is it possible to directly write the entire (on and off-diagonal)
pressure tensor or viscosity from amber md_simulation? If I need to edit
some source code then how to do it?
2. Is there any documentation that clearly shows how to calculate the
pressure tensor or viscosity by post-processing from amber velocity forces
and trajectory? If anyone did something like this then also please help me
by sharing how you did it.
Regards
Newaz MD Fahim
MEXT Scholar
WPI-NanoLSI
Kanazawa University
[image: image.png]
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
