Date: Mon, 10 Feb 2025 12:46:07 +0000
Dear AMBER Support Team,
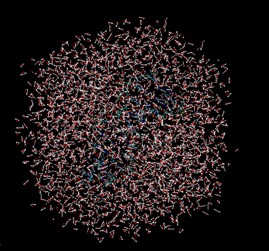
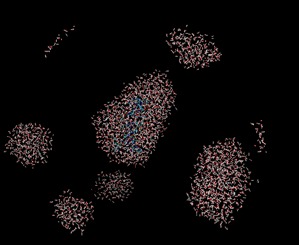
I hope this email finds you well. I have been running MD simulations using AMBER on a CPU and have obtained good results. However, when I shift to CUDA (GPU) simulations, I am encountering the following issues:
1. The water box appears broken in VMD.
2. Some water molecules cluster together unnaturally.
3. The overall system visualization differs significantly from the CPU-generated trajectory.
I have followed the same input parameters and input files for both CPU and GPU runs using examples in amber tutorial. My simulation setup includes:
• Software: AMBER22
• Force Field: [ff14SB + TIP3P]
• Processor: 13th gen i9 Intel 13900k
Cuda NVIDIA GPU RTX4090
I would appreciate any insights or troubleshooting steps you can suggest. Please let me know if you need additional information or input files. I have attached two images one for CUDA and one from CPU.
Looking forward to your response.
Best regards,
Tushar Gupta
[Image]
With Sander CPU
[Image]
With CUDA GPU
Sent from Outlook for Android<https://aka.ms/AAb9ysg>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: IMG-20250210-WA0012.jpg)
(image/jpeg attachment: IMG-20250210-WA0013.jpg)
