Date: Wed, 11 Dec 2024 16:22:33 +0000
Hi Dr. Giese, I've included Shi Zhang from Prof. York's group in this email. I'm not sure if Shi is the right person to contact. I appreciate your assistance. If not, Shi, please disregard this message.
I encountered some issues when calculating relative binding free energy using Amber TI and Dr. Giese told me that my simulation had poor phase space overlap.
Is there any way I can improve the phase space overlap? Because of the poor overlap, when using edgembar, it generated py file containing "inf" and python .py failed.
Currently, I'm using one-stage (unified) TI with 21 windows (not ACES, I found ACES took a really long time for equilibration and TI) to calculate the relative binding free energy.
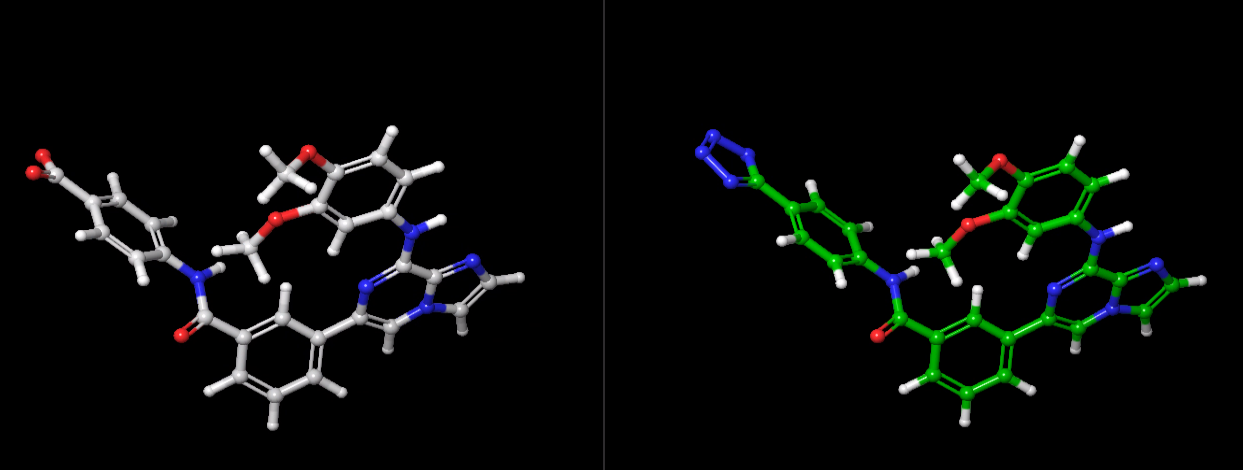
Dr. Giese helped me to make the two attached phase space overlap histograms between neighboring states. Is the poor overlap associated with the large softcore size (see the attached structures, the benzene disappeared in the second ligand) or bad mdin settings (attached ti.in)? More detailed output files are uploaded to [https://res-1.cdn.office.net/assets/mail/file-icon/png/folder_16x16.png] AMBER TI<https://purdue0-my.sharepoint.com/:f:/g/personal/li728_purdue_edu/EoGs5diEcXdLu52kfb_tttUBYKjf_TqHaIYVNb4Rl1n9RQ?e=gu1QGT>
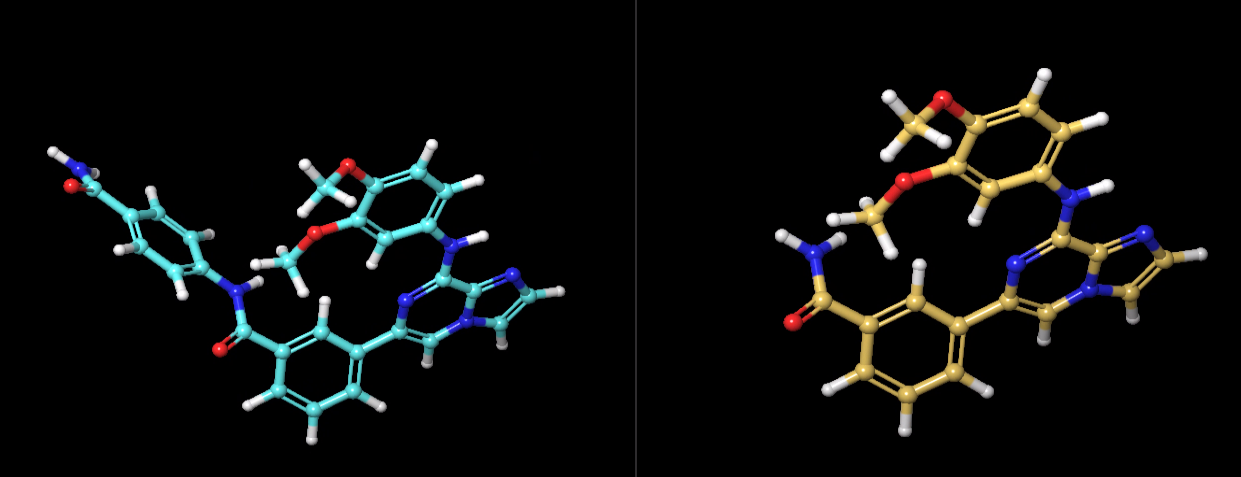
Dr. Giese, I simulated additional two ligands with smaller size of softcore (3 atoms transformed to 5 atoms), shown below. I still have poor overlap for complex and still cannot use edgembar to generate correct py file. I think these two ligands are pretty similar, do you think I should extend the simulation time for each window to longer than 6 ns or I need to switch to ACES?
Thanks for your help.
Regards,
Tongtong
[cid:eaa6647c-b23f-46f9-9734-3b94ab2b3965][cid:ca60f4cb-b28f-4b56-9815-6c5aea82b591][cid:d604ba4d-9dc6-413b-8b4f-a3a85303898b]
________________________________
From: Timothy Giese <giese025.rutgers.edu>
Sent: Tuesday, December 10, 2024 7:26 PM
To: Tongtong Li <li728.purdue.edu>; AMBER Mailing List <amber.ambermd.org>
Subject: Re: amber TI edgembar | py generated from xml contains inf
---- External Email: Use caution with attachments, links, or sharing data ----
Thank you for providing your data. I looked at it today, and the main results are summarized below.
The smoothstep softcore lambda scheduling is designed to enforce dU/dlambda = 0 at lambda=0 and lambda=1. This does not mean you ignore the dU/dlambda; it only means you exactly know the value (you still have to integrate from lambda=0 to lambda=1 — you shouldn't integrate from lambda=0.05 to lambda=0.95).
1.
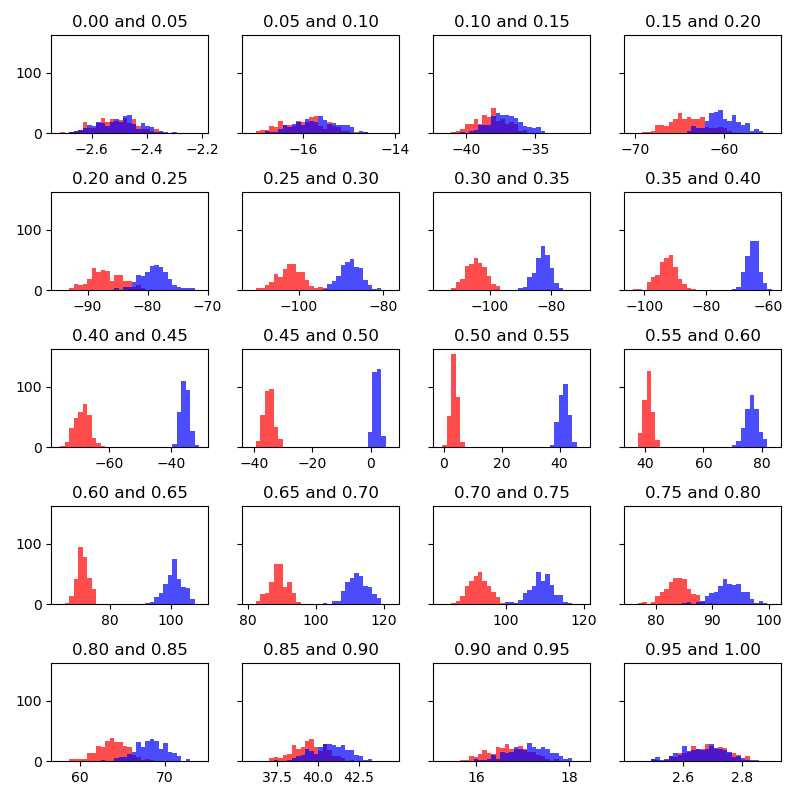
I attached the histograms corresponding to the energy differences from neighboring states. I attached a script used to produce the plots. In the complexed environment, there are many neighboring states with poor phase space overlap (gaps of 40 kcal/mol between the histograms). Therefore, one should expect FEP-like methods to complete fail in this case. I can't recall any other system that has been shown to me that exhibits large gaps like this. It makes me question what is so different between this system and the RBFE networks people in our group have been working on.
2.
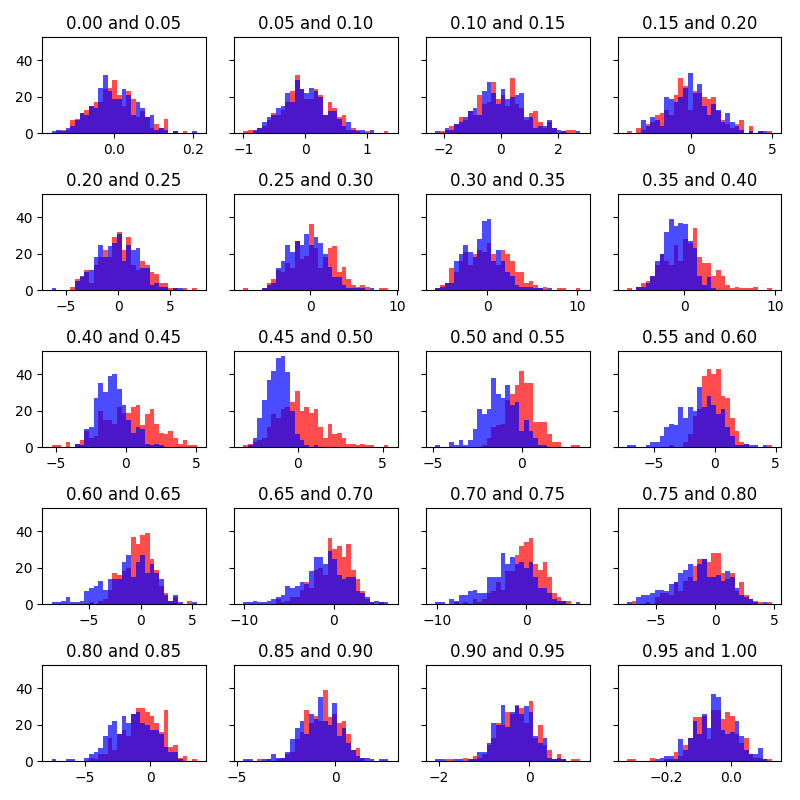
I calculated the solvated environment dG values from your data with "edgembar --no-auto" and compared it to the result produced from the pymbar program. I verified that both programs yield the exact same dG value using the BAR method, and both programs similarly yield the same dG value using the MBAR method.
3.
The solvated environment dG values produced by TI, BAR, and MBAR do not agree with each other, however. I haven't determined why — TBH I haven't seen a situation where all 3 methods produce very different values, but it must have something to do with the data that is being produced rather than the analysis considering the agreement between multiple independent analysis programs. Maybe one of Prof York's students could try running the simulations with their free energy workflow to see if they can reproduce the symptom with the latest development code, or perhaps they might notice something wrong with the system or trajectories by visual inspection. I wonder if the problem (disagreement between the 3 methods) goes away when a smaller softcore region is used? -- I'm not sure what hidden limitations exist with the GPU implementation regarding softcore spatial size, number of softcore atoms, or other features.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
(image/png attachment: complex.png)

(image/png attachment: solvated.png)
- text/plain attachment: AllHist.py.txt

(image/png attachment: 05-complex.png)
(image/png attachment: 06-solvated.png)
- application/octet-stream attachment: ti.in
(image/png attachment: Screenshot_2024-12-11_104108.png)

{kind=link}