Date: Tue, 27 Aug 2024 17:12:03 +0000
Hi all,
I'm just reaching out today to ask if it is possible to reorganize the atom
order of an AMBER trajectory file to match that of a specific topology
file. At the moment, I'm trying to perform backbone principle component
analysis, however in my modified nucleotide simulation trajectory and
topology files compared those of my unmodified nucleotide simulation,
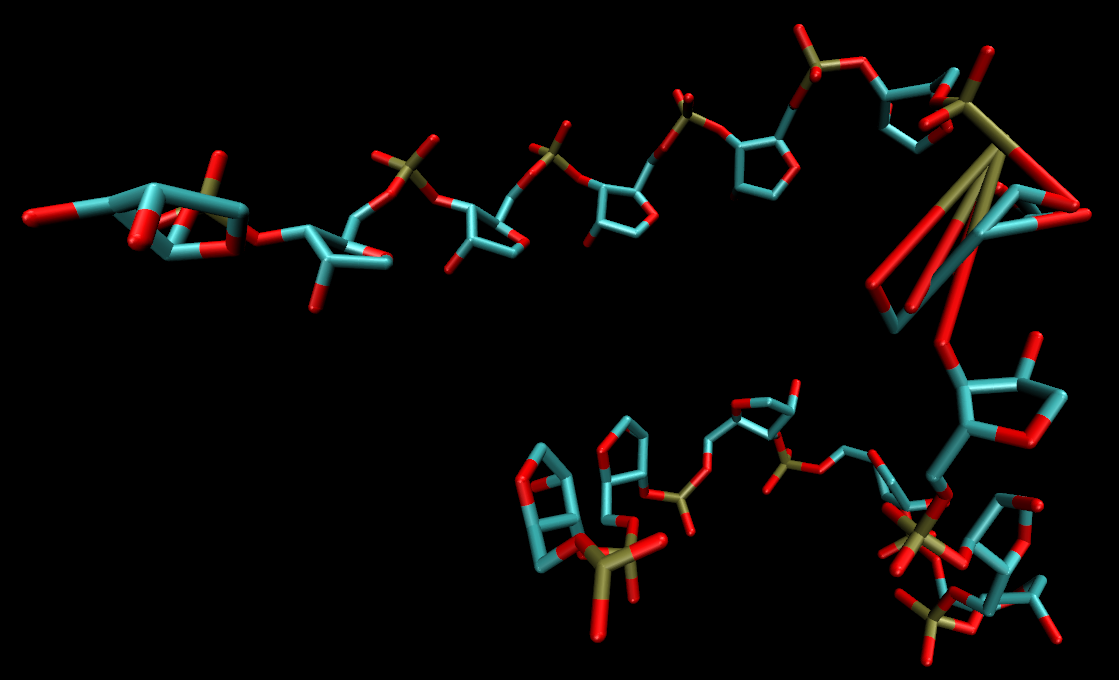
there's a discrepancy between the ribose atom orders. Due to this atom
ordering discrepancy, when I load my modified trajectory file into my
unmodified topology file, the connectivity at the ribose of my modification
is completely awry. Are there any commands offered by cpptraj that can help
re-order my trajectory files? Or am I out of luck? Attached is also a
screenshot of the faulty ribose connectivity I referenced.[image:
d087bf61-7cc9-456c-ad02-388370f290d1.png]
I feel as though the "remap" command may work for my case, however I'm also
unfamiliar with this command's syntax so any help would be greatly
appreciated.
V. Best,
Kyle Warren
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: d087bf61-7cc9-456c-ad02-388370f290d1.png)
