Date: Fri, 10 May 2024 08:43:40 +0000
Dear all Amber users and Experts,
applying the ZAFF Modeling Tutorial
for the Center ID 4 I am experimenting
many errors with tleap.
In the metal center type box of the previous
Center ID there is written:
"Zn-CCHH ( with two C as CYM ,one H as HID"
It means that is applicable when there are
two HID or one HID and one HIE?
In the associate example , 1A1F , all the
three ZFs have two HID.
I have used this method for a protein of my
interest where one of the three ZFs has
two CYM , one HID and one HIE.
The content of tleap_ZAFF.in is:
source leaprc.protein.ff14SB
source leaprc.water.tip3p
addAtomTypes { { "ZN" "Zn" "sp3" } { "S4" "S" "sp3" } { "N3" "N" "sp3" } }
loadamberparams frcmod.ions1lm_126_tip3p
loadamberprep ZAFF.prep
loadamberparams ZAFF.frcmod
mol = loadpdb example_ZAFF.pdb
# ZF: HIS 256 CYS 260 HIS 263 CYS 278 ZN 526
bond mol.526.ZN mol.256.NE2
bond mol.526.ZN mol.260.SG
bond mol.526.ZN mol.263.ND1
bond mol.526.ZN mol.278.SG
savepdb mol example_ZAFF_dry.pdb
saveamberparm mol example_ZAFF_dry.prmtop example_ZAFF_dry.inpcrd
solvatebox mol TIP3PBOX 10.0
charge mol
addions mol CL 0
savepdb mol example_ZAFF_solv.pdb
saveamberparm mol example_ZAFF_solv.prmtop example_ZAFF_solv.inpcrd
quit
Some of the errors that I receive are:
.....................................
Building topology.
Building atom parameters.
Building bond parameters.
/home/xxxx/sources/amber22/bin/teLeap: Error!
Could not find bond parameter for atom types: NA - ZN
for atom ND1 at position -7.302631, 8.675131, -3.184384
and atom ZN at position -6.086631, 10.026131, -2.860384.
Building angle parameters.
/home/xxxx/sources/amber22/bin/teLeap: Error!
Could not find angle parameter for atom types: N3 - ZN - NA
for atom NE2 at position -6.231631, 10.901131, -1.293384,
atom ZN at position -6.086631, 10.026131, -2.860384,
and atom ND1 at position -7.302631, 8.675131, -3.184384.
/home/xxxx/sources/amber22/bin/teLeap: Error!
Could not find angle parameter for atom types: S4 - ZN - NA
for atom SG at position -6.747631, 11.568131, -4.450384,
atom ZN at position -6.086631, 10.026131, -2.860384,
and atom ND1 at position -7.302631, 8.675131, -3.184384.
..................................................................
/home/xxxx/sources/amber22/bin/teLeap: Error!
** No torsion terms for atom types: H-NA-ZN-S4
for atom HD1 at position -6.687973, 9.320285, -2.708914,
atom ND1 at position -7.302631, 8.675131, -3.184384,
atom ZN at position -6.086631, 10.026131, -2.860384,
and atom SG at position -3.887631, 9.186131, -2.955384.
/home/xxxx/sources/amber22/bin/teLeap: Error!
** No torsion terms for atom types: NA-ZN-S4-CT
for atom ND1 at position -7.302631, 8.675131, -3.184384,
atom ZN at position -6.086631, 10.026131, -2.860384,
atom SG at position -3.887631, 9.186131, -2.955384,
and atom CB at position -3.872631, 7.777131, -1.812384.
/home/xxxx/sources/amber22/bin/teLeap: Error!
** No torsion terms for atom types: CC-NA-ZN-S4
for atom CG at position -8.689631, 8.520131, -3.043384,
atom ND1 at position -7.302631, 8.675131, -3.184384,
atom ZN at position -6.086631, 10.026131, -2.860384,
and atom SG at position -3.887631, 9.186131, -2.955384.
............................................................................................
Where I'm wrong?
Thanks.
Saverio
P.S.
The complete log is in tleap_1.log
example_ZAFF.pdb is the the used pdb.
image_ZF.png is the image of the ZF that should be modeled.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- text/x-log attachment: tleap_1.log
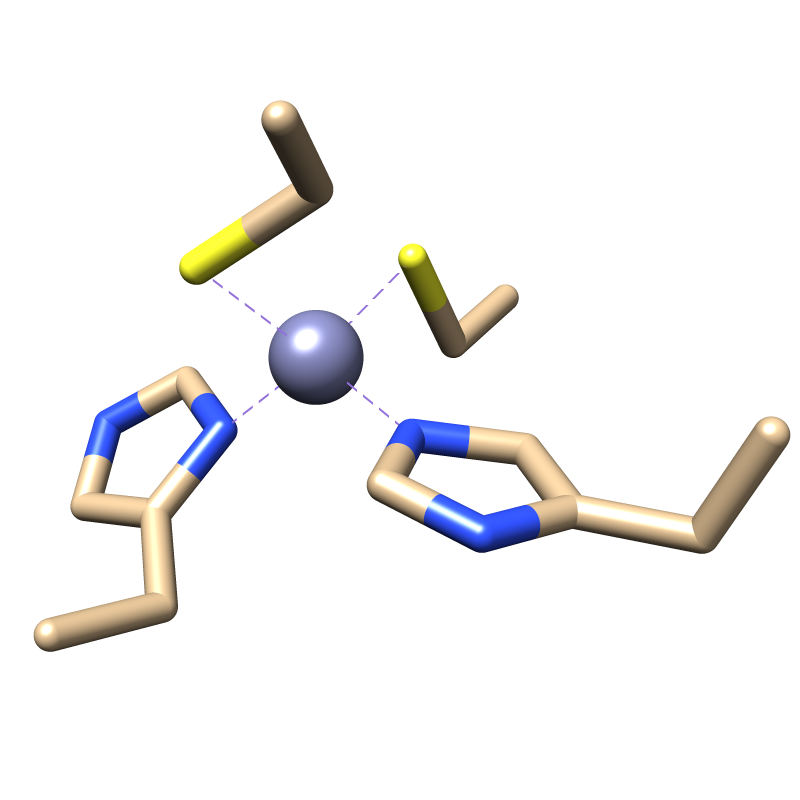
(image/png attachment: image_ZF.png)
- chemical/x-pdb attachment: example_ZAFF.pdb
