Date: Mon, 31 Jul 2023 11:38:13 +0200
Dear Amber users, Maria and Gerardo!
Thank you for your help, I finally managed to run my simulation, but it
wasn't so easy to fix the issue.
One thing was that I was missing restraints on the ATP-Mg complex, and also
my dihe.restraint file was wrong and I needed to adjust it as well.
Luckily, a colleague reached me and gave me his working ATP-Mg complex
parameters.
But I'm still wondering about the parameterization of the ATP-Mg complex. I
built a system in CHARMM-GUI, and I wanted to run the simulations using two
different engines: gromacs and Amber. For gromacs, the CHARMM-GUI files
were working just fine, but for Amber they weren't. And the most important
change I did was changing the ATP structure and parameters.
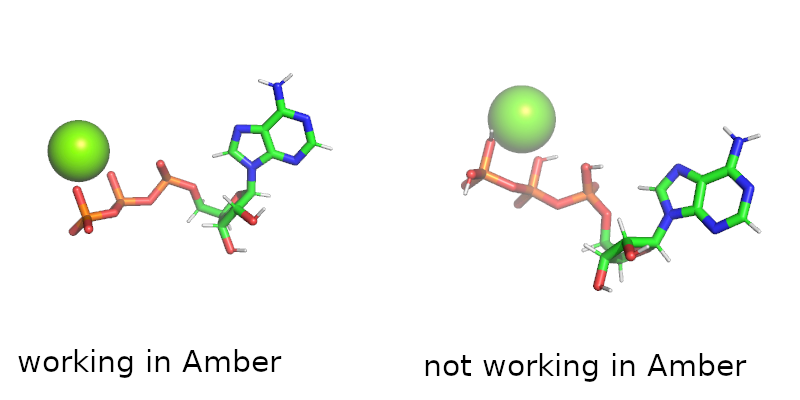
In the gromacs system the ATP I simulated contains hydrogens in the
phosphorus groups, while in the system for Amber - it did not. I'm
attaching a figure to show you what I mean.
Also, because of that, I needed to add 8 K+ ions in the system to
neutralize the charge, and now the systems are different for each
simulation engine.
Is there another way to parameterize the ATP-Mg complex here? Because I
would like to have the same number of ions in the system for both Amber and
gromacs simulations.
I would be glad for any idea on how to parameterize the complex to keep the
hydrogens!
Best regards,
Karolina
czw., 20 lip 2023 o 14:50 Gerardo Zerbetto De Palma <g.zerbetto.gmail.com>
napisał(a):
> Hello Karolina.
> Have you tried a minimization run on that system? I have had problems with
> CHARMM-GUI inputs in the past, especially with the dihedral restraints, so
> I make my own minimization-equilibration inputs. I usually use a
> modification of the equilibration protocol that can be found in the AMBER
> tutorial for membrane proteins:
> https://ambermd.org/tutorials/advanced/tutorial16/index.php#Molecular_Dynamics
> <https://ambermd.org/tutorials/advanced/tutorial16/index.php#Molecular_Dynamics>
> In case you need more hints on this, just write back.
> Regards!
> Gera!
>
> El jue, 20 jul 2023 a las 9:12, Karolina Mitusińska (Markowska) via AMBER
> (<amber.ambermd.org>) escribió:
>
>> Dear Amber users,
>>
>> I need your help in setting up a membrane-bound protein which also
>> contains
>> an ATP-Mg complex in its structure.
>> I prepared the input files using CHARMM-GUI, but when I try to run a
>> simulation using those files, the system is blowing up during the first
>> step of equilibration.
>> After few blow-ups I managed to successfully run an MD simulation of the
>> whole protein, membrane, water and ions, but without the ATP-Mg complex. I
>> got a short production run in which I did not see anything weird. So I
>> believe that the MD simulation crashes because of the ATP parameters I'm
>> using.
>>
>> However, when I used the ATP.frcmod file I got from CHARMM-GUI and used
>> the
>> pdb file containing the ATP complex and Mg ion, I managed to run the
>> simulation as well; I got the trajectories from a production run.
>>
>> I am attaching my .mdout file from the failed equilibration.
>> I could really need some help in getting this system running.
>>
>> Looking forward to your help.
>> Best, Karolina
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>>
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: atp_question.png)
