Date: Wed, 28 Jun 2023 14:16:41 +0900
Hi,
I performed a 1000 step energy minimization of a small organic molecule,
which is prepared by antechamber with GAFF2 and the RESP-derived charges.
While the final configuration after minimization looks fine, but there
were occasional huge bumps in the potential energy in the course of the
minimization (I attached the energy plot file and the mdout file).
I looked the trajectory and confirmed that the configurations were also
distorted at the timing of the energy bumps.
Has anyone observed any situation like this? What may be the cause of
this behavior?
# software version: Amber22/AmberTools22
# Example of the energy bump (line 3952-3997 of the attached min.out
file. The corresponding trajectory snapshots are also attached.) :
NSTEP ENERGY RMS GMAX NAME NUMBER
479 4.2420E+01 5.0983E-02 1.3987E-01 C11 15
BOND = 1.7474 ANGLE = 14.0204 DIHED = 9.6009
VDWAALS = -3.2458 EEL = -60.2337 HBOND = 0.0000
1-4 VDW = 6.5164 1-4 EEL = 74.0143 RESTRAINT =
0.0000
NSTEP ENERGY RMS GMAX NAME NUMBER
480 1.4703E+04 1.5718E+04 6.7010E+04 C11 15
BOND = 1990.7664 ANGLE = 737.5050 DIHED = 53.9941
VDWAALS = -0.8177 EEL = -62.7268 HBOND = 0.0000
1-4 VDW = 11889.5108 1-4 EEL = 95.2040 RESTRAINT =
0.0000
NSTEP ENERGY RMS GMAX NAME NUMBER
481 9.9038E+02 2.5920E+02 9.5956E+02 C11 15
BOND = 561.1639 ANGLE = 338.1107 DIHED = 21.0169
VDWAALS = -2.3347 EEL = -61.6524 HBOND = 0.0000
1-4 VDW = 53.2071 1-4 EEL = 80.8659 RESTRAINT =
0.0000
NSTEP ENERGY RMS GMAX NAME NUMBER
482 4.2420E+01 3.8712E-02 1.1602E-01 O1 11
BOND = 1.7456 ANGLE = 14.0208 DIHED = 9.6006
VDWAALS = -3.2456 EEL = -60.2342 HBOND = 0.0000
1-4 VDW = 6.5169 1-4 EEL = 74.0157 RESTRAINT =
0.0000
Best Regards,
Kotaro
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
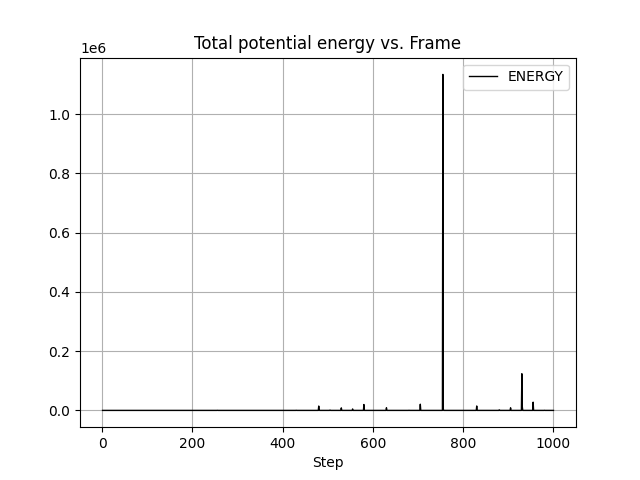
(image/png attachment: 230628_energy.png)
- text/plain attachment: min.out
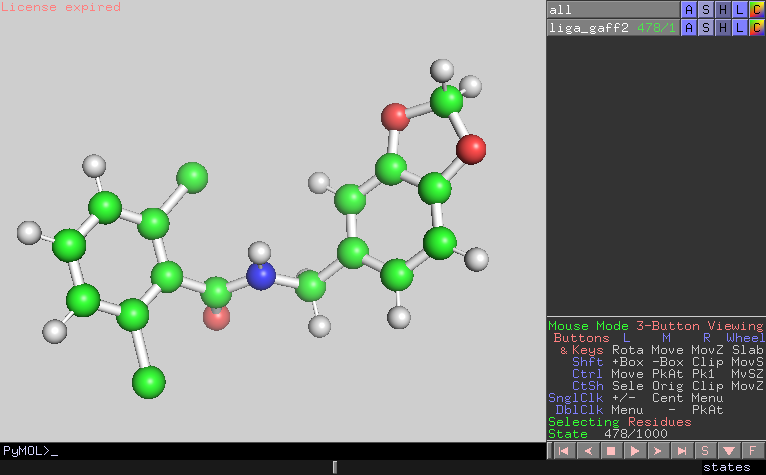
(image/png attachment: frame478.png)
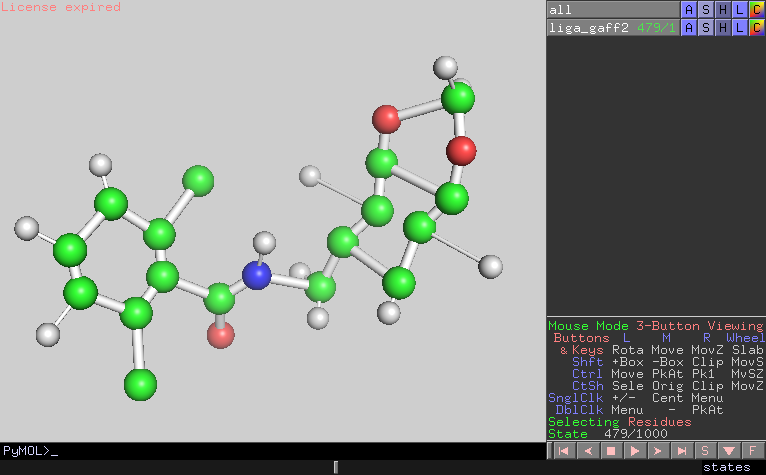
(image/png attachment: frame479.png)
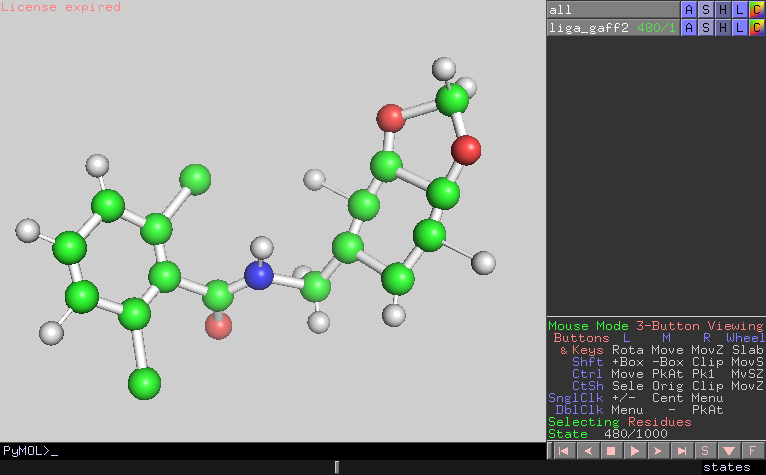
(image/png attachment: frame480.png)
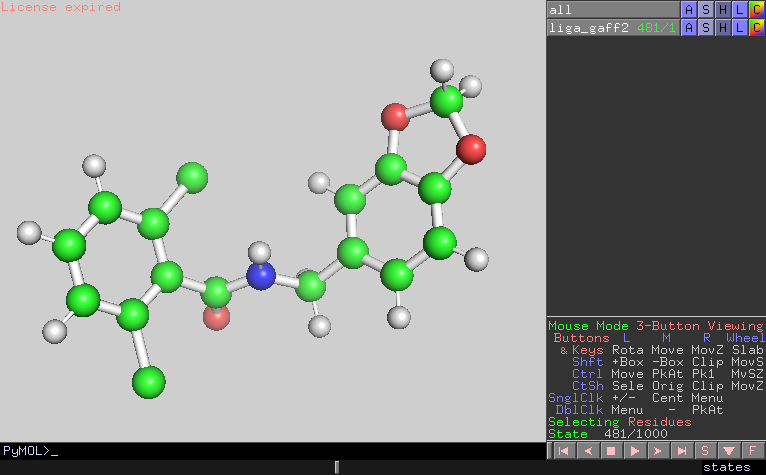
(image/png attachment: frame481.png)
