Date: Wed, 5 Apr 2023 15:29:25 -0700
Hi Fanyu,
Regarding the need for some form of correction for charge changing
mutations during alchemical transformation, I will refer to this nice
paper: https://pubs.acs.org/doi/10.1021/acs.jctc.8b00825
The observation of ions overlapping with softcore regions (especially with
introducing or destroying charged regions i.e. aspartic acid) is sometimes
referred to as "particle collapse". There are also a few literature
references that note this (
https://www.nature.com/articles/s41598-022-14443-z). Our understanding is
that this is due to van der waals repulsions becoming too weak and
electrostatic attraction leading to atom overlap. One workaround can be to
separate the van der waals transformation from the
charging/decharging transformations.
In Amber22, I believe new softcore potentials were introduced to help
mitigate this "particle collapse" and to better enable charge/van der waals
transformations in a single transformation. However, I have not had a
chance to test these myself.
Michael
On Mon, Apr 3, 2023 at 6:49 PM Fanyu Zhao <fz2113.nyu.edu> wrote:
> Hi Michael,
>
> Thank you so much for your explanation. It's really helpful!
>
> I have successfully gained the dual-topology file with a Na+
> transferring to WAT. I realized that Na+ and WAT were not necessarily part
> of atoms that needs to be merged, so I just merged the common part of
> protein and then add Na+ and WAT to the soft core in TI simulation.
>
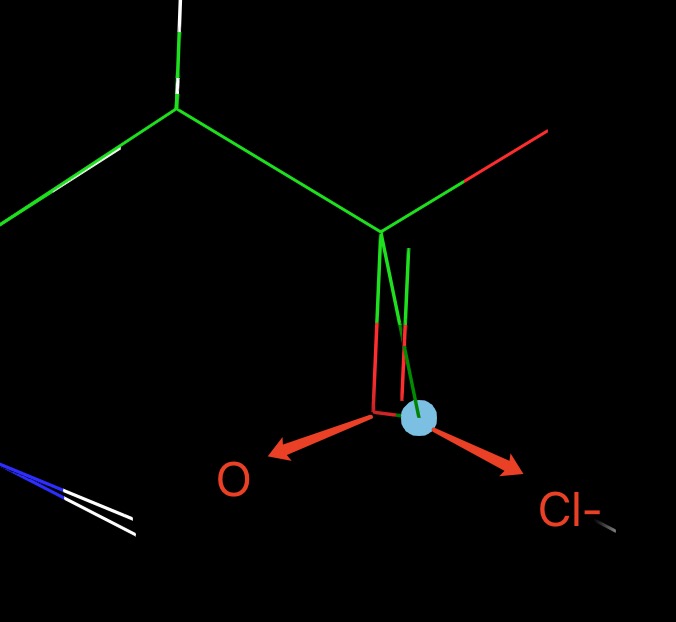
> But I encountered another problem that the Na+ in sc will overlap with a
> random atom (same for the Cl-) and this will cause an error in TI. This
> will happen even though I put a restraint on Na+ and WAT in sc. A picture
> is attached.
>
> The keyword setting for example is timask1=":3, 386", timask2=":12,396",
> scmask1=":3, 386", scmask2=":12,396". Res 3 and 12 are amino acids; res 386
> is Na+ and res 396 is WAT.
>
> I was also wondering if I could just run TI simulation without any
> change to Na+ or WAT when the wild type and mutated amino acids with
> charge-change are involved, since AMBER will add a neutralizing plasma
> around the system.
>
> Thank you so much for your help!
>
> Best,
> Fanyu
>
> [image: image.png]
>
> On Tue, Apr 4, 2023 at 1:30 AM Michael T Kim <micke.kim.gmail.com> wrote:
>
>> Hi Fanyu,
>>
>> To my understanding, timerge is a means of improving calculation
>> efficiency (by not duplicating calculations for common atoms). The biggest
>> efficiency gains for your proposed simulation is probably by merging the
>> common protein atoms since these are ~100 residues. I wouldn't worry about
>> the WAT and Na+ in timerge. Note: in amber20, we also experienced trouble
>> with timerge when we were trying to merge topologies where the mutated
>> amino acid wasn't part of the last two protein chains. Workaround was to
>> reorder the protein chains so that the chains that have the mutations are
>> the last two protein chains (i.e. I would reorder your pdb so that :22-100
>> comes first, then :1-10, then :11-20, then sodium and waters)
>>
>> For the WAT to Na+ transformation, have you tried the following:
>>
>> - make sure the oxygen on water and Na+ have the same coordinates.
>> Include the oxygen and Na+ in the timask (but not scmask) so that their
>> coordinates are linked
>> - include the hydrogens on water in the scmask
>>
>> mike
>>
>> On Mon, Mar 27, 2023 at 1:14 AM Fanyu Zhao via AMBER <amber.ambermd.org>
>> wrote:
>>
>>> Dear Amber users and developers,
>>>
>>> I have a question about how to prepare dual-topology file for TI
>>> simulation when converting charge is involved in the calculation of
>>> relative binding free energy of protein mutation.
>>>
>>> For instance, I'm now trying to mutate an Asp to Ala, which means the
>>> charge of the TI region is converted from +1 to 0. I find that in some
>>> paper, the authors will simultaneously change a WAT to Na+ when mutating
>>> the amino acids.
>>>
>>> I was wondering how I can get the dual-topology structure with the above
>>> method if I'm using timerge module in Parmed. It always gives me error
>>> messages that I can only merge adjacent residues. Because the WAT lines
>>> will always be put at the end of the pdb file including both wild-type
>>> and
>>> mutant protein. It's always very far away from the wild type, mutants,
>>> and
>>> chosen Na+ I'm going to merge.
>>>
>>> For instance, residue :1-10 is TI region 1 (chain with Asp), :11-20 is TI
>>> region 2 (chain with Ala), :21 is Na+ (will disappear in the end state),
>>> :22-100 are other chains, :101-200 is WAT. The WAT I choose to appear in
>>> the end state is residule :101 that is definitely not adjacent to res
>>> :1-21.
>>>
>>> Thank you for taking the time to read this email!
>>>
>>> Best,
>>> Fanyu
>>> _______________________________________________
>>> AMBER mailing list
>>> AMBER.ambermd.org
>>> http://lists.ambermd.org/mailman/listinfo/amber
>>> <https://urldefense.proofpoint.com/v2/url?u=http-3A__lists.ambermd.org_mailman_listinfo_amber&d=DwMFaQ&c=slrrB7dE8n7gBJbeO0g-IQ&r=TA6Yv1GqGslhLaifgggoUQ&m=Vzz2yxQO9JVQGWFnog6YlL114z-pS6AQvmBViUqylswzrrU7Ba7mxIBgaJ1KS1S9&s=Ll28yk8T_XmajXhp20iDzSN9ROfYe8mlR98-7GPVz9E&e=>
>>>
>>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
