Date: Thu, 5 Jan 2023 05:35:33 +0000
Hello Dear fellow amber users,
I have a question regarding preparation of a non-standard KCX residue that has a charge -1. I helped myself using two tutorials https://ambermd.org/tutorials/advanced/tutorial1/section2.htm and http://ambermd.org/tutorials/basic/tutorial5/
I began my journey with drawing the KCX residue with a charge -1 and ACE and NME caps on each side of the residue there the protien continues. I performed a MMFF94 geometry optimization to get a 3D structure, saved it as -mol2 file and used OpenBabel to convert it to .xyz file. Then I performed a ligand optimization and electrostatic grid calculation of the ligand with Gaussian 16.
Optimization:
%NProcShared=8
%chk=opt_geom.chk
%mem=4000MB
#p opt HF/6-31G(d) nosymm iop(6/7=3) gfinput
Ligand optimization
-1 1
N 0.91855 -0.24842 -0.05187
C 0.22768 0.85908 -0.70521
C -0.66191 0.32043 -1.85691
O -1.13106 -0.81503 -1.86175
C 2.77243 -1.75052 0.36762
C -0.60772 1.66221 0.30755
C 0.27282 2.31308 1.38789
C -0.53354 3.15093 2.38690
C 0.38019 3.78389 3.44261
N -0.33255 4.59174 4.39467
C -0.93530 4.07714 5.44751
O -0.78185 2.85174 5.67876
O -1.59215 4.94164 6.08269
N -0.91449 1.25855 -2.84175
C -1.71657 0.98174 -4.00873
C 2.19197 -0.60772 -0.42101
O 2.83372 -0.03308 -1.29552
H 0.34013 -0.93422 0.41863
H 0.97890 1.52039 -1.15197
H -1.35011 1.00836 0.78408
H -1.17052 2.44232 -0.21929
H 1.02449 2.94890 0.90407
H 0.81630 1.53592 1.93842
H -1.29121 2.52443 2.87118
H -1.07206 3.94408 1.85340
H 0.98211 3.03203 3.96609
H 1.08745 4.45825 2.94564
H -0.51215 5.56228 4.20415
H -0.44067 2.15193 -2.79430
H -2.58875 1.64031 -3.98586
H -1.12232 1.21573 -4.89488
H -2.04191 -0.06071 -4.05186
H 2.98814 -2.58311 -0.30728
H 3.69884 -1.41310 0.84197
H 2.09284 -2.09440 1.15132
Using a command g16 < /filename.com/ > /filename.out/
El. grid calculation:
%NProcShared=8
%chk=opt_geom.chk
%mem=600MB
#p HF/6-31G(d) nosymm iop(6/33=2,6/41=10,6/42=17) pop=mk guess=read gfinput geom=check
Ligand electrostatic grid calculation
-1 1
Using a command g16 < /el_grid.com/ > /el_grid.out/
Next, I used antechamber:
> antechamber -i el_grid.out -fi gout -bk KCX -fo ac -o KCX.ac -c bcc -at amber -nc -1 -m 1 -rn KCX
Then I created a file KCX_1.mc
I labeled HEAL_NAME and TAIL_NAME, MAIN_CHAIN of all of the heavy atoms that KCX consists of, OMIT_NAME contains the atoms of ACE and NME caps, PRE_HEAD_TYPE, POST_TAIL_TYPE and CHARGE. See below:
HEAD_NAME N1
TAIL_NAME C2
MAIN_CHAIN C1
MAIN_CHAIN O1
MAIN_CHAIN C4
MAIN_CHAIN C5
MAIN_CHAIN C6
MAIN_CHAIN C7
MAIN_CHAIN N2
MAIN_CHAIN C8
MAIN_CHAIN O2
MAIN_CHAIN O3
OMIT_NAME C10
OMIT_NAME O4
OMIT_NAME C3
OMIT_NAME H16
OMIT_NAME H17
OMIT_NAME H18
OMIT_NAME N3
OMIT_NAME H12
OMIT_NAME C9
OMIT_NAME H13
OMIT_NAME H14
OMIT_NAME H15
PRE_HEAD_TYPE C
POST_TAIL_TYPE N
CHARGE -1.00
Now I run the script using prepgen:
> prepgen -i KCX.ac -o KCX_1.prepin -m KCX_1.mc -rn KCX
Everything is fine and I get no error so far.
Then I continue with parmchk2 and create two frcmod files one with amber and one with gaff parameters as tutorial 5 recomends:
> parmchk2 -i KCX_1.prepin -f prepi -o KCX_1.frcmod -a Y -p $AMBERHOME/dat/leap/parm/parm10.dat
> grep -v "ATTN" KCX_1.frcmod > KCX_2.frcmod # Strip out ATTN lines
> parmchk2 -i KCX_1.prepin -f prepi -o KCX_3.frcmod
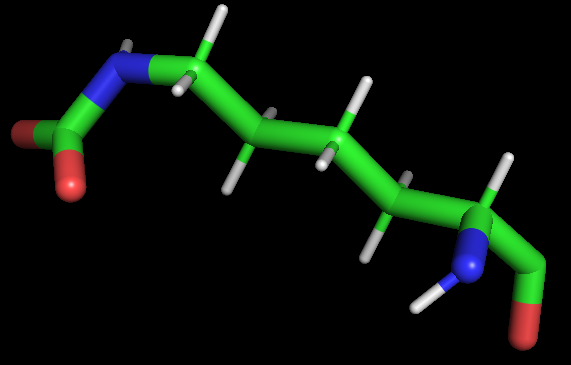
I also checked the NEWPDB.PDB file and it looks good - like a KCX residue should. There are two double bonds between C and O atoms missing in this representation however.
[Inline image OWAPstImg969435]
Then I prepare leap.in file as follows:
source leaprc.gaff2
source leaprc.protein.ff14SB
source leaprc.water.tip3p
set default PBRadii mbondi3
loadamberprep KCX_1.prepin
loadamberparams KCX_3.frcmod
loadamberparams KCX_2.frcmod
a = loadPDB 2jff_OK2.pdb
addIons a Na+ 0
addIons a Cl- 0
solvateBox a TIP3PBOX 10 iso
savepdb a system_leap_3.pdb
saveamberparm a system_3.top system_3.coord
quit
And I run it using:
> tleap -f leap.in
Results:
I get ZERO errors, however I get 10 warnings (more in leap1.log file).
/apps/amber/amber20/bin/teLeap: Warning!
addIons: 1st Ion & target unit have charges of the same sign:
unit charge = -2e-06; ion1 charge = -1;
can't neutralize.
/apps/amber/amber20/bin/teLeap: Warning!
Converting N-terminal residue name to PDB format: NALA -> ALA
/apps/amber/amber20/bin/teLeap: Warning!
Converting C-terminal residue name to PDB format: CARG -> ARG
/apps/amber/amber20/bin/teLeap: Warning!
Converting N-terminal residue name to PDB format: NARG -> ARG
/apps/amber/amber20/bin/teLeap: Warning!
Converting C-terminal residue name to PDB format: CGLN -> GLN
/apps/amber/amber20/bin/teLeap: Warning!
Converting N-terminal residue name to PDB format: NGLU -> GLU
/apps/amber/amber20/bin/teLeap: Warning!
Converting C-terminal residue name to PDB format: CHID -> HID
>
> saveamberparm a system_3.top system_3.coord
Checking Unit.
/apps/amber/amber20/bin/teLeap: Warning!
There is a bond of 8.716 angstroms between O3 and C2 atoms:
------- .R<KCX 198>.A<O3 22> and .R<KCX 198>.A<C2 23>
/apps/amber/amber20/bin/teLeap: Warning!
There is a bond of 3.202 angstroms between O1 and C4 atoms:
------- .R<KCX 198>.A<O1 5> and .R<KCX 198>.A<C4 6>
Building topology.
Building atom parameters.
Building bond parameters.
Building angle parameters.
Building proper torsion parameters.
Building improper torsion parameters.
** Warning: No sp2 improper torsion term for O2-CT-C-N
atoms are: O3 C1 C2 N
old PREP-specified impropers:
<KCX 198>: -M C1 N1 H1
<KCX 198>: C8 C7 N2 H11
<KCX 198>: N2 O2 C8 O3
<KCX 198>: C1 +M C2 O1
total 1274 improper torsions applied
4 improper torsions in old prep form
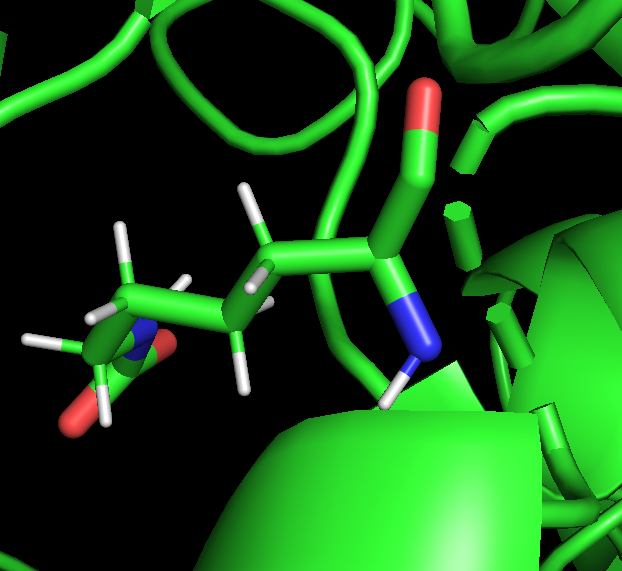
When I look the .pdb created by the leap.in, it doesn0t look like the KCX residue is connected to the protein since we can see the dotted line where a residue should be in a cartoon vizualization.
[Inline image OWAPstImg263658]
I additionally created a restart file.
When I run minization nvt.in script containing:
n water nvt restraint
&cntrl
imin = 0, irest =0, ntx = 1, ntxo = 1, ioutfm = 1,
ntb = 1, pres0 = 1.0, ntp = 0,
taup = 2.0,
cut = 10, ntr = 1,
ntc = 2, ntf = 2,
tempi = 0.0, temp0 = 50.0,
ntt = 3, ig = -1, gamma_ln = 1.0,
nstlim = 10000, dt = 0.0005,
ntpr = 1, ntwx = 5000, ntwr = 5000,
vlimit=20
/
Hold the protein fixed
100.0
RES 1 433
END
END
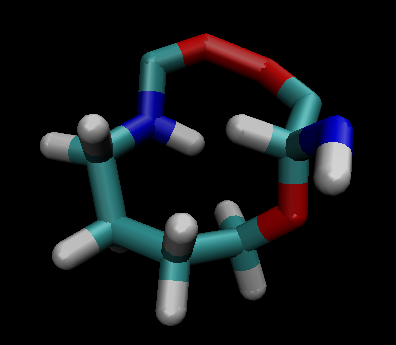
When I vizualize the first .nc file created it somehow forms wierd bonds and connects the residue in a loop.
[Inline image OWAPstImg604716]
Now I'm wondering where have I failed with the creation of non-standard residue. And now I have two questions:
1. Regarding the circle that the residue created
Is it the .mc file? How should it be written differently? Is it something else completely?
2. Regarding the lack of bond created
Should I have written in leap.in file for the bond to be created?
bond a./number_of_residue1/.C a./number_of_residue2/.N
bond a./number_of_residue2/.N a./number_of_residue3/.C
Please advise me, I no longer see what else could I have done differently. Thank you
Barbara
Barbara Herlah
Doktorska študentka / Young Researcher
[cid:image001.png.01D69D88.0294FD40]
[cid:image002.png.01D69D88.0294FD40]
Teoretični odsek / Theory Department
Kemijski inštitut / National Institute of Chemistry
Hajdrihova 19
SI-1000 Ljubljana
Slovenija / Slovenia
00386 1 47 60 394
barbara.herlah.ki.si www.ki.si<https://www.ki.si/>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: OutlookEmoji-Inline_image_OWAPstImg969435ecb07e49-a105-4870-83cd-c8593fec4580.png)
(image/png attachment: OutlookEmoji-Inline_image_OWAPstImg263658efbcd7ce-0bb7-429f-b4a0-52403cb6e8ce.png)
(image/png attachment: OutlookEmoji-Inline_image_OWAPstImg604716642bb317-4448-49cb-affd-52a9925ef4e6.png)
- application/octet-stream attachment: leap1.log
