Date: Thu, 27 Oct 2022 12:23:06 +0800 (GMT+08:00)
您好:
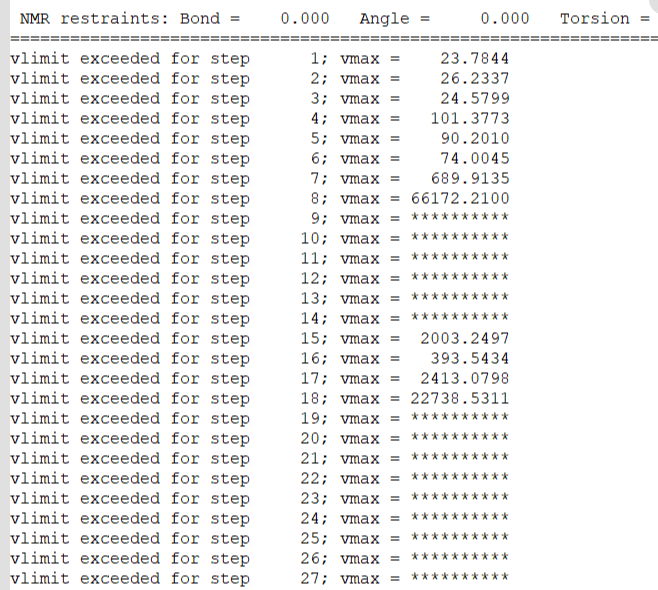
有个问题想请教一下,我在超算集群环境中使用ambertools22的sander.MPI命令计算文件,输出结果中发现:
如果用少量核数运行,比如 mpirun -np 32 sander.MPI -ng 8 -groupfile ./groupfile ,可以得到正常的计算结果
但是如果增加运行的核数,比如 mpirun -np 64 sander.MPI -ng 8 -groupfile ./groupfile,输出的output文件,会说体系内原子的速度异常,数值太高,显示为一堆星号,算到最后值从一堆星号变为NaN
使用的计算环境和文件都相同,唯一的区别是计算核数不同。
请问一下造成这样问题的原因可能是什么呢?
感谢!
丁帅威
技术服务工程师
并行科技
联系我
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
