Date: Thu, 30 Jun 2022 16:46:51 +0000
Dear AMBER maniacs,
I am simulating a box containing a protein P in a mix of solvents S1 and S2 in presence of an electrolyte EL using AMBER20 on GPU acceleration.
I want to calculate the radial distribution function of EL molecules around P and their relative coordination numbers.
When I use the following command in cpptraj:
radial out OutputFileName.dat 0.1 15 :EL_ResidueMask :P_ResidueMask noimage volume bymol1 byres2
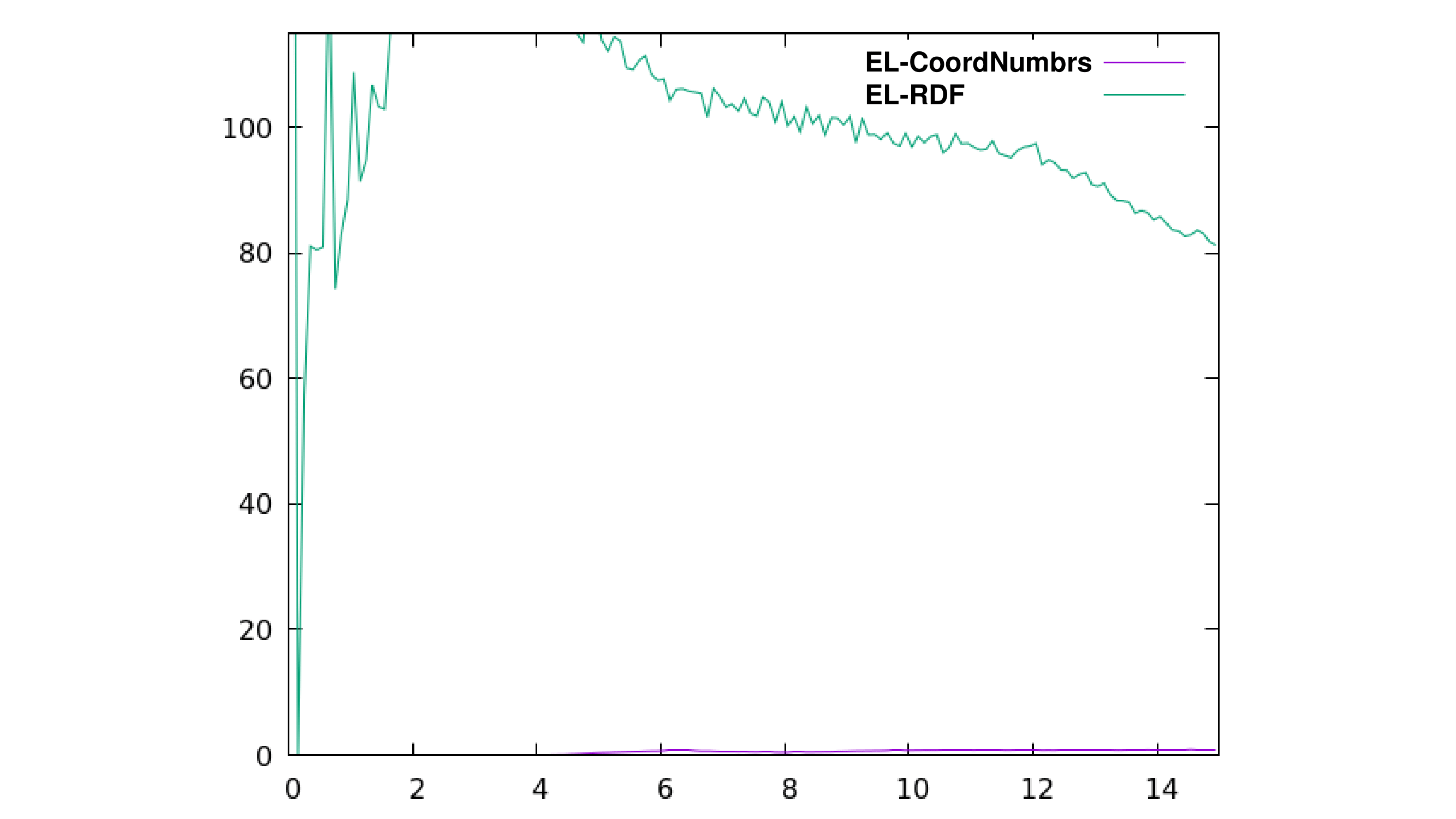
it retrieves what I think is the RDF of EL molecules around P calculated by using the density of the whole box in each frame, taking into account EL molecules and protein residues but please correct me if I am wrong. When plotted, the RDF looks like the green curve in the attached image.
When I use the command:
radial out OutputFileName.dat 0.1 15 :EL_ResidueMask :P_ResidueMask noimage volume center1 center2
I get what I think are the coordination numbers between EL molecules and P by using the density of the whole box in each frame, considering the interaction between the center of both EL molecules and P but the values range seems to be quite small to me as if the 1st solvation shell for EL was recorded at 6nm distance from P and it accounted for 1 molecule (represented in purple in the image). I didn't use any normalisation so I'd expected similar values. Is this wrong to think?
Or have I just swapped the two commands?
I also wanted to know that since from the theory we know that the relation between RDF and coordination numbers between two particles a and b as a function of the distance r is:
N{ab}(r) = rho G{ab}(r)
where rho is the density and G(r) the RDF, what is the keyword that differentiate between RDF to coordination numbers in the commands I used?
Is it right to use the "volume" keyword if I want to calculate the properties of the electrolyte in the mix of solvents or should I have used "density" along with the value for density retrieved by
0.6022/MEL (with MEL the mass of an EL molecule)?
Kind regards,
Damiano
This message and any attachment are intended solely for the addressee
and may contain confidential information. If you have received this
message in error, please contact the sender and delete the email and
attachment.
Any views or opinions expressed by the author of this email do not
necessarily reflect the views of the University of Nottingham. Email
communications with the University of Nottingham may be monitored
where permitted by law.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: RDFvsCN-1.png)
