Date: Mon, 20 Jun 2022 15:09:29 -0400
Dear Amber Community,
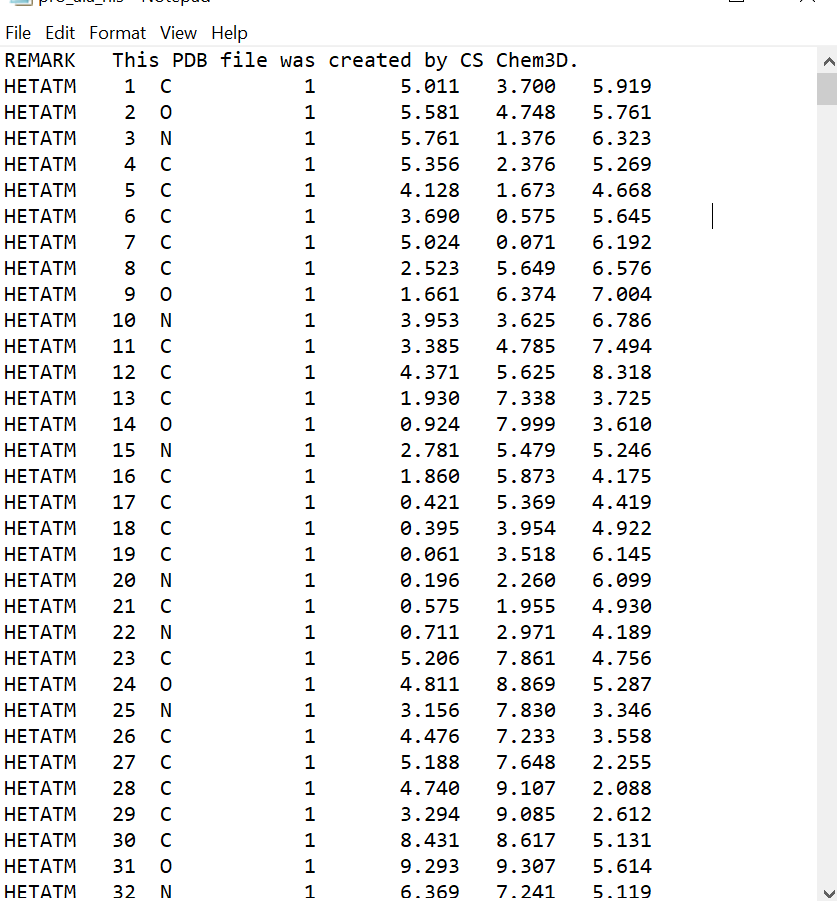
I am having trouble preparing my pdb protein for Amber.
I start by drawing my peptide sequence on ChemDraw and download it as a
.pdb file. When I open that pdb file on a text editor app, I replace any
"HETATM" with "ATOM" and delete any "CONNECT" lines. When I type in the
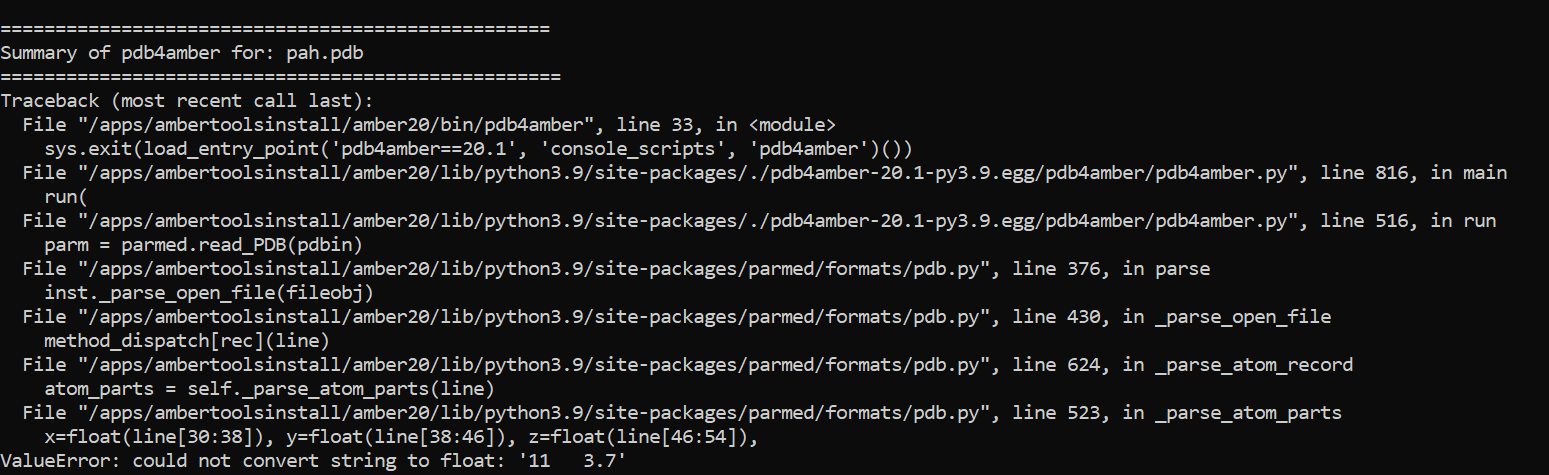
pdb4amber command however, it gives me an error message saying
==================================================
Summary of pdb4amber for: pah.pdb
===================================================
Traceback (most recent call last):
File "/apps/ambertoolsinstall/amber20/bin/pdb4amber", line 33, in <module>
sys.exit(load_entry_point('pdb4amber==20.1', 'console_scripts',
'pdb4amber')())
File
"/apps/ambertoolsinstall/amber20/lib/python3.9/site-packages/./pdb4amber-20.1-py3.9.egg/pdb4amber/pdb4amber.py",
line 816, in main
run(
File
"/apps/ambertoolsinstall/amber20/lib/python3.9/site-packages/./pdb4amber-20.1-py3.9.egg/pdb4amber/pdb4amber.py",
line 516, in run
parm = parmed.read_PDB(pdbin)
File
"/apps/ambertoolsinstall/amber20/lib/python3.9/site-packages/parmed/formats/pdb.py",
line 376, in parse
inst._parse_open_file(fileobj)
File
"/apps/ambertoolsinstall/amber20/lib/python3.9/site-packages/parmed/formats/pdb.py",
line 430, in _parse_open_file
method_dispatch[rec](line)
File
"/apps/ambertoolsinstall/amber20/lib/python3.9/site-packages/parmed/formats/pdb.py",
line 624, in _parse_atom_record
atom_parts = self._parse_atom_parts(line)
File
"/apps/ambertoolsinstall/amber20/lib/python3.9/site-packages/parmed/formats/pdb.py",
line 523, in _parse_atom_parts
x=float(line[30:38]), y=float(line[38:46]), z=float(line[46:54]),
ValueError: could not convert string to float: '11 3.7'
I've included screenshots below of a portion of my .pdb file and the error
message.
Any insight would be appreciated.
Thanks,
Beatriz Goncalves
[image: image.png]
[image: image.png]
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
(image/png attachment: 02-image.png)
