Date: Thu, 10 Feb 2022 10:52:19 -0300
Dear amber users
I'm facing a problem in a protein-binding simulation
I'm trying to simulate 100ns of molecular dynamics.
I made the coupling and later the Dynamic process
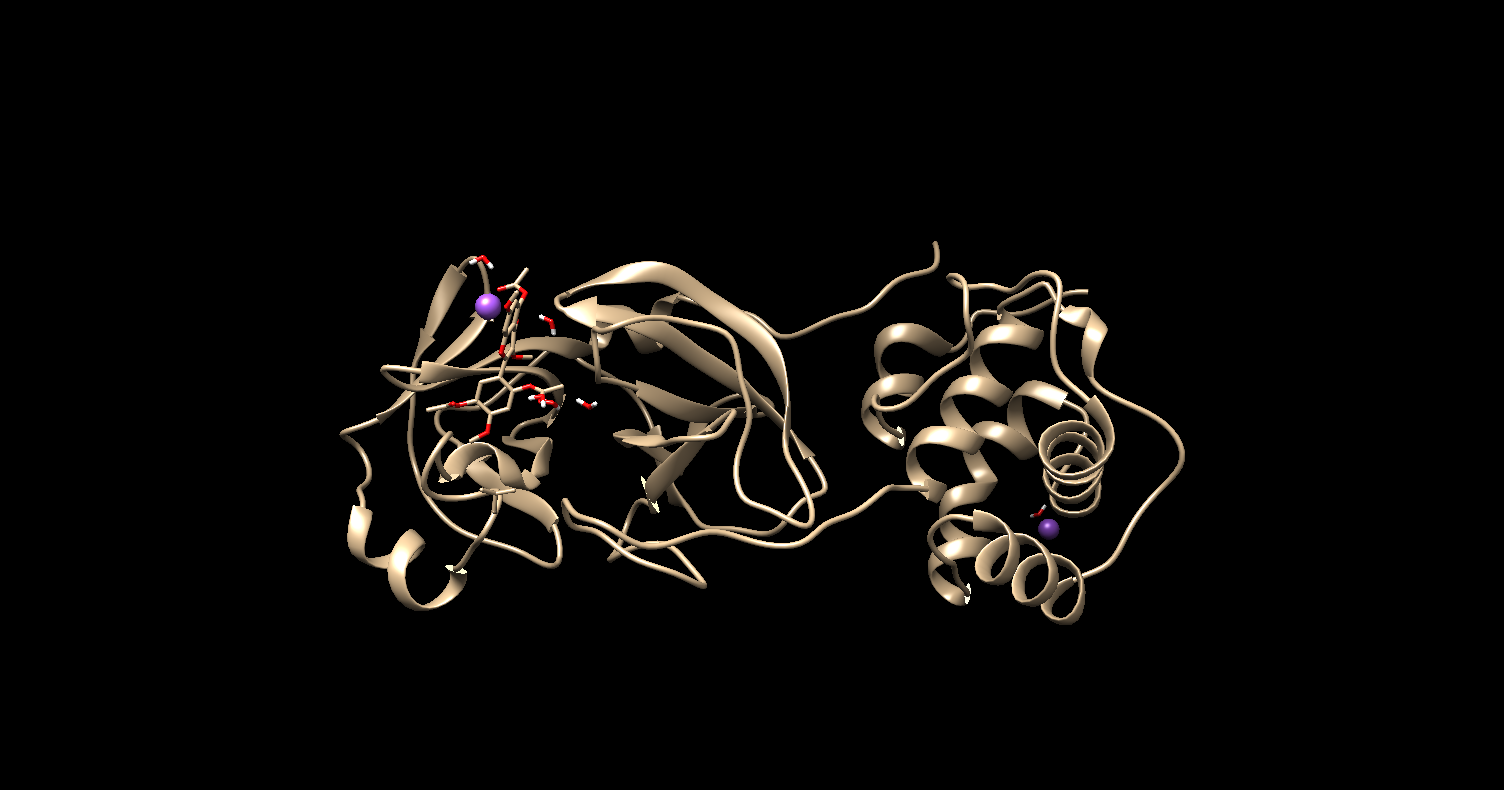
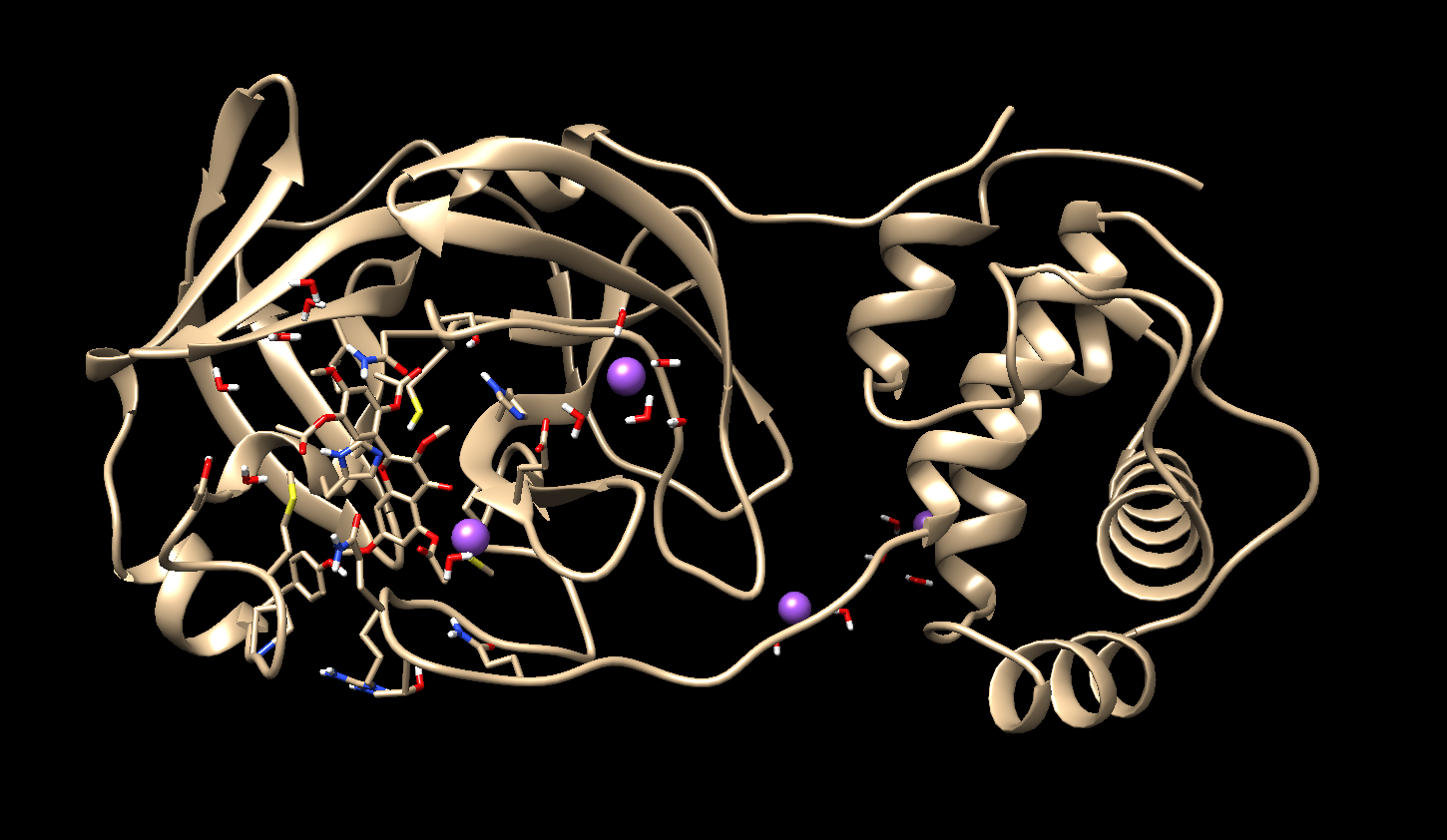
1) In the initial 40ns, my ligand moves away from the binding site and
consequently from the enzyme.
What can I do to solve this problem?
2) When preparing the system in tleap, I notice that a Na+ is placed very
close to my ligand and remains there during the entire simulation time, is
this a problem? if yes how to solve?
follow the image
thanks
-- Prof Dr Renato Costa Instituto Federal do Pará - IFPA Grupo de Modelagem Molecular - UFPA Tel.+55 91 985484622
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Captura_de_ecr___de_2022-02-10_10-50-51.png)
(image/png attachment: Captura_de_ecr___de_2022-02-10_10-51-30.png)
