Date: Thu, 12 Aug 2021 00:49:11 -0300
dear amber users
I'm trying to run a molecular diamic simulation with the SARS-CoV-2 Spike
Protein protein.
In preparing my system I'm using:
source leaprc.protein.ff14SB
source leaprc.gaff2
source leaprc.water.tip3p
loadamberparams frcmod.ionsjc_tip3p
set default PBRadii mbondi2
loadamberparams lig_resp.frcmod
loadoff ON.lib
rec = loadpdb pro.pdb
LIG = loadmol2 lig_resp.mol2
com = combine {rec ON}
savepdb ON binding.pdb
saveamberparm ON ligand.prmtop ligand.inpcrd
savepdb rec receiver.pdb
saveamberparm rec receiver.prmtop receiver.inpcrd
savepdb with complex.pdb
saveamberparm with complex.prmtop complex.inpcrd
cartoon with
additions with Na+ 0
additions with Cl- 0
cartoon with
solvateoct with TIP3PBOX 20.0
savepdb with complex_solv.pdb
saveamberparm with complex_solv.prmtop complex_solv.inpcrd
quit
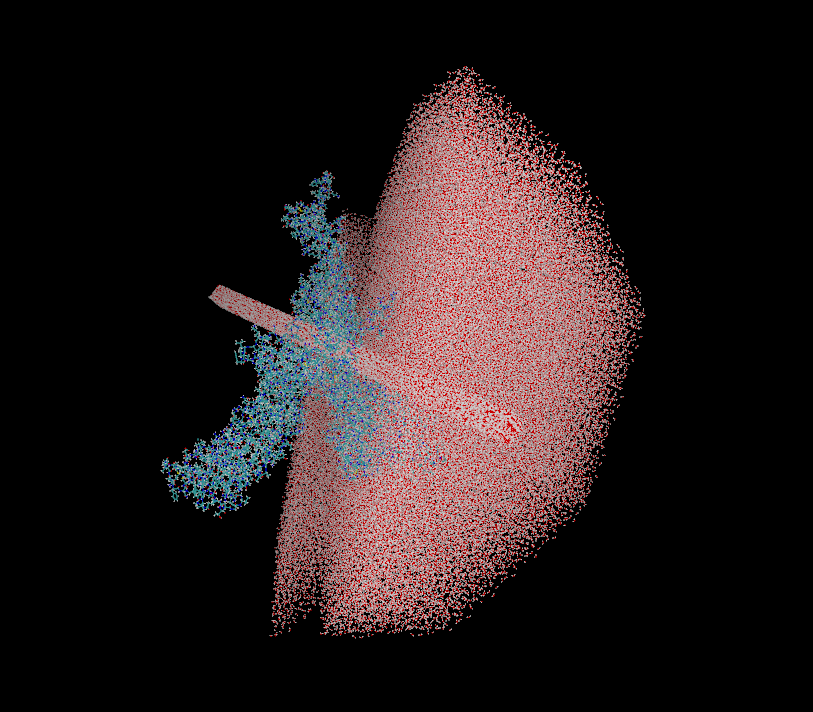
When I open the system in the VMD I have the following image attached.
Could someone tell me what mistake I'm making so that my complex doesn't
get into the water tank?
Thanks
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Captura_de_ecr___de_2021-08-12_00-48-42.png)
